Chapter 21 Enols and Enolates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Ochem ACS Review 18 Enols and Enolates
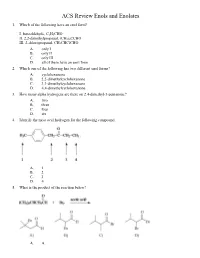
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Lecture 6 the Crossed Aldol Reaction and Its Many Variants
Lecture 6 The Crossed Aldol Reaction and its Many Variants Objectives: By the end of this lecture you will be able to: 1. make an appropriate choice of base to completely enolise carbonyl compounds; 2. use enolates in a crossed aldol reaction; 3. recognise the aldol functional group motif, and its variants, in complex molecules; 4. use the aldol disconnection to simplify a retrosynthetic analysis; 5. use the Reformatski and Mukaiyama aldol reactions in synthesis; 6. draw arrow-pushing mechanisms for all the aldol reactions discussed in this lecture. Introduction We have seen how choosing a base (B-) whose conjugate acid (B−H) is a much poorer acid − i.e. has a much higher pKa − than the proton we wish to abstract, ensures effectively complete deprotonation of the α-C−H of a carbonyl compound to provide the corresponding enolate. By completely enolising the carbonyl group, we can suppress self-condensation aldol processes, and instead use a different electrophile such as an aldehyde to form a β-hydroxy carbonyl compound. This reaction sequence is identical in basic mechanism to the intramolecular aldol processes that we discussed in previous lectures. It is now described as a crossed aldol condensation because the electrophile is a different carbonyl compound to the one that was used to form the nucleophilic enolate. This reaction, and its many variants, provides one of the most important methods for preparing C−C bonds. Pattern Recognition The aldol reaction and its many variants are very useful reactions in synthesis. You need to be able to identify the patterns or functional group motifs where this type of bond-forming process can be used. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -
20.3 Alkylation of Enolate Anions
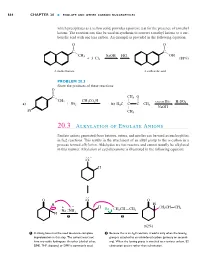
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

MINDO-Forces Study on the Substituent Effect in the Keto-Enol Tautomerism of Acetyl Derivatives
MINDO-Forces Study on the Substituent Effect in the Keto-Enol Tautomerism of Acetyl Derivatives Wasim F. Al-Halasah, Ali Mahasnah, and Salim M. Khalil Chemistry Department, College of Science, University of Mutah, Karak, Jordan Reprint requests to Prof. S.M.K.; E-mail: [email protected] Z. Naturforsch. 59a, 299 – 308 (2004); received February 11, 2004 MINDO-Forces calculations with complete geometry optimization have been performed on ac- etaldehyde, vinyl alcohol and acetyl derivatives CH3COX(X=H, F, OH, CN, NH2,NO2,CH3, CF3,OCH3). It was found that acetaldehyde is more stable than vinyl alcohol by 10.451 kcal/mol. Thermodynamically, keto tautomers are more stable than their enol counterparts. This agrees with theoretical calculations. The electron releasing substituents tend to stabilize keto tautomers, while the electron withdrawing substituents tend to destabilize the keto tautomers, relative to the parent. Geometrical parameters, heats of formation, electron densities, Gibbs free energies and orbital ener- gies (HOMO-LUMO) are reported. Key words: Acetaldehyde; Vinyl Alcohol; Keto-Enol Tautomerism; Acetyl Derivative. 1. Introduction 311++G∗∗//MP2(full)/6-31G∗ levels of theory, ac- etaldehyde 1 is found to lie 11.2 and 13.35 kcal/mol Tautomerism refers to the equilibrium between two below vinyl alcohol 2 on the potential energy sur- different structures of the same compound. It is a pro- face, respectively. Recent B3LYP and G2MP2 calcu- totropic rearrangement in which a hydrogen at the α- lations [14] show that acetaldehyde 1 lies 10.4 and position to a carbon-heteroatom double bond migrates 11.1 kcal/mol below vinyl alcohol 2, respectively. -

The Claisen Condensation
Lecture 19 The Claisen Condensation •• •• • • O • O • – • CH3COCH2CH3 • CH2 COCH2CH3 March 29, 2016 Chemistry 328N Exam Tomorrow Evening!! Review tonight 5PM -6PM Welch 1.316 Chemistry 328N Some “loose ends” before we go on Spectrosopy of acid derivatives A selective reduction for your tool box Chemistry 328N Reduction of Acid Derivatives Acids (page 679-681) Esters (page 738-739) Please work through the example on 738 Amides (page 739-742) Nitriles (page 742) Selective reductions with NaBH4 Chemistry 328N DIBAlH Diisobutylaluminum hydride (DIBAlH) at -78°C selectively reduces esters to aldehydes •at -78°C, the tetrahedral intermediate does not collapse and it is not until hydrolysis in aqueous acid that the carbonyl group of the aldehyde is liberated Stable at low temperature Chemistry 328N Infrared Spectroscopy C=O stretching frequency depends on whether the compound is an acyl chloride, anhydride, ester, or amide. C=O stretching frequency n O O O O O CH3CCl CH3COCCH3 CH3COCH3 CH3CNH2 1822 cm-1 1748 1736 cm-1 1694 cm-1 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Anhydrides have two peaks due to C=O stretching. One from symmetrical stretching of the C=O and the other from an antisymmetrical stretch. C=O stretching frequency n O O CH3COCCH3 1748 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Nitriles are readily identified by absorption due to carbon-nitrogen triple bond stretching that is “all alone” in the 2210-2260 cm-1 region. Chemistry 328N Hydrolysis and Decarboxylation Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl ester hydrolysis Note which bond is broken in this hydrolysis !! Chemistry 328N Recall our discussion of the acidity of protons a to carbonyls The anion is stabilized by resonance The better the stabilization, the more acidic the a proton Acidity of a protons on“normal” aldehydes and ketones is about that of alcohols and less than water…pKa ~ 18-20 Some are far more acidic, i.e. -

The Claisen Condensation Is a Carbon–Carbon Bond Forming Reaction That Occurs Between Two Esters Or One Ester and Another Carb
Claisen Ester Condensation The Claisen condensation is a carbon–carbon bond forming reaction that occurs between two esters or one ester and another carbonyl compound in the presence of a strong base, resulting in a β-keto ester or a β-diketone. Requirements ❖ At least one of the reagents must be enolizable (have an α-proton and be able to undergo deprotonation to form the enolate anion). ❖ There are a number of different combinations of enolizable and nonenolizable carbonyl compounds that form a few different types of Claisen. ❖ The base used must not interfere with the reaction by undergoing nucleophilic substitution or addition with a carbonyl carbon. ❖ For this reason, the conjugate sodium alkoxide base of the alcohol formed (e.g. sodium ethoxide if ethanol is formed) is often used, since the alkoxide is regenerated. ❖ In mixed Claisen condensations, a non-nucleophilic base such as lithium diisopropylamide, or LDA, may be used, since only one compound is enolizable. ❖ LDA is not commonly used in the classic Claisen or Dieckmann condensations due to enolization of the electrophilic ester. ❖ The alkoxy portion of the ester must be a relatively good leaving group. ❖ Methyl and ethyl esters, which yields methoxide and ethoxide, respectively, are commonly used. Types Mechanism ❖ In the first step of the mechanism, an α-proton is removed by a strong base, resulting in the formation of an enolate anion, which is made relatively stable by the delocalization of electrons. ❖ Next, the carbonyl carbon of the (other) ester is nucleophilically attacked by the enolate anion. ❖ The alkoxy group is then eliminated (resulting in (re)generation of the alkoxide), and the alkoxide removes the newly formed doubly α-proton to form a new, highly resonance-stabilized enolate anion. -

CHEM 330 Topics Discussed on Sept. 18 Principle: a Claisen Condensation Promoted by Nah/Cat. Etoh Takes Place So That All Steps
CHEM 330 Topics Discussed on Sept. 18 Principle: a Claisen condensation promoted by NaH/cat. EtOH takes place so that all steps but the final deprotonation of EtOH by NaH are reversible. Consequently, the system will tend to attain a thermodynamic energy minimum, and the product that forms will be the thermodynamically most favorable one. A reaction occurring under such conditions is said to proceed under thermodynamic control Principle: mixing one equivalent of pure ethyl acetoacetate with excess pure ethanol in the presence of a catalytic amount of EtONa will induce a reverse Claisen condensation ("retro-Claisen"), resulting in conversion of the starting acetoacetate into two molecules of ethyl acetate (the thermodynamically more favorable state of the system): – CH3-CO-CH2-COOEt + EtOH —[ cat. EtO ]—> 2 CH3-COOEt (overall ΔG < 0) Principle of microscopic reversibility: in each step of any given transformation, the forward and the reverse reactions occur along the same mechanistic pathway; i.e., by identical mechanisms operating in reverse Example: a reverse Claisen condensation occurs as follows: O O O O Na + Na + H–OEt OEt OEt OEt pKa ≈ 12 H pKa ≈ 17 + H–OEt + H–OEt 5 Keq ≈ 10 although the above equilibrium is shifted to the right, there will always be some residual free EtO and ethyl acetoacetate in the medium. Under the usual approximations (CHEM 203): –5 –3 [CH3COCH2COOEt] = [EtO ] ≈ 10 ≈ 3•10 – These residual concentrations of EtO( ) and CH3COCH2COOEt are sufficient to induce a slow, but inexorable, reverse reaction. O O O O O O + Na OEt OEt OEt OEt OEt OEt + H–OEt + H–OEt Na + H–OEt O O OEt OEt OEt Na H notice that the reversible reaction depends on proton exchanges among the various species: NaH removes all such protons and suppresses the retro-Claisen process lecture of Sept 18 p. -
Enolates, Enols and Enamines
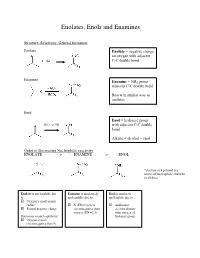
Enolates, Enols and Enamines Structure, Reactivity, General formation Enolate Enolate = negative charge on oxygen with adjacent C-C double bond Enamine Enamine = NR2 group adjacent C-C double bond Reacts in similar way as enolates Enol Enol = hydroxyl group - H3O+ or OH with adjacent C-C double bond Alkene + alcohol = enol Order of Decreasing Nucleophilic reactivity ENOLATE > ENAMINE > ENOL *electron-rich pi bond is a source of nucleophilic character in all three Enolate is nucleophilic due Enamine is moderately Enol is moderate to… nucleophilic due to… nucleophile due to… Oxygen’s small atomic radius N (EN=3) is less Additional Formal negative charge electronegative than electron density oxygen (EN =3.5) from oxygen of Detractors to nucleophilicity: hydroxyl group Oxygen is more electronegative than N When looking at which side is favored (products or reactants): Equilibrium favors side with WEAKER acid/base pair Weakest acid and weakest base should be on SAME side When comparing pKa values, juxtapose just the acids or just the bases Side with larger pka value (weaker acid) favored in equilibrium Remember that differences in pKa values reflect a difference quantity of 10x Example: compound H-A (pKa=3) + base compound A (pKa=10) + H-base 7 equilibrium lies to the RIGHT by factor of 10 Some Useful pKa values (from more acidic basic) H2SO4 (pka = -9) H2O (pka = 15.7) + H3O (pka = -1.8) CH3COCH3 (pka = 19) B-diketone (pka = 9) CH3COOCH3 (pKa = 25) HCN (pka = 9.1) LDA-H (pka = 36) CH3OH (pka = 15.5) How do we know which proton to deprotonate? Deprotonate most acidic proton usually leads to the most stable enolate 1. -
Claisen Condensation – Beyond Labz Virtual Chemlab Activity Purpose: 1) to Examine How Choice of Reagent and Solvent Impacts the Outcome of a Claisen Condensation
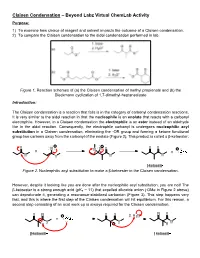
Claisen Condensation – Beyond Labz Virtual ChemLab Activity Purpose: 1) To examine how choice of reagent and solvent impacts the outcome of a Claisen condensation. 2) To compare the Claisen condensation to the aldol condensation performed in lab. Figure 1. Reaction schemes of (a) the Claisen condensation of methyl propionate and (b) the Dieckmann cyclization of 1,7-dimethyl-heptanedioate Introduction: The Claisen condensation is a reaction that falls is in the category of carbonyl condensation reactions. It is very similar to the aldol reaction in that the nucleophile is an enolate that reacts with a carbonyl electrophile. However, in a Claisen condensation the electrophile is an ester instead of an aldehyde like in the aldol reaction. Consequently, the electrophile carbonyl is undergoes nucleophilic acyl substitution in a Claisen condensation, eliminating the -OR group and forming a ketone functional group two carbons away from the carbonyl of the enolate (Figure 2). This product is called a β-ketoester. Figure 2. Nucleophilic acyl substitution to make a β-ketoester in the Claisen condensation. However, despite it looking like you are done after the nucleophilic acyl substitution, you are not! The - β-ketoester is a strong enough acid (pKa ~ 11) that expelled alkoxide anion ( OMe in Figure 2 above) can deprotonate it, generating a resonance-stabilized carbanion (Figure 3). This step happens very fast, and this is where the first step of the Claisen condensation will hit equilibrium. For this reason, a second step consisting of an acid work-up is always required for the Claisen condensation. Figure 3. Deprotonation and subsequent acid work-up to generate the final, neutral β-ketoester product in the Claisen condensation.