Lecture 6 the Crossed Aldol Reaction and Its Many Variants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
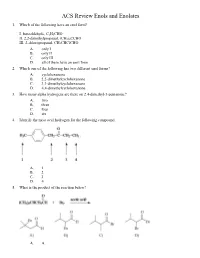
Ochem ACS Review 18 Enols and Enolates
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

MINDO-Forces Study on the Substituent Effect in the Keto-Enol Tautomerism of Acetyl Derivatives
MINDO-Forces Study on the Substituent Effect in the Keto-Enol Tautomerism of Acetyl Derivatives Wasim F. Al-Halasah, Ali Mahasnah, and Salim M. Khalil Chemistry Department, College of Science, University of Mutah, Karak, Jordan Reprint requests to Prof. S.M.K.; E-mail: [email protected] Z. Naturforsch. 59a, 299 – 308 (2004); received February 11, 2004 MINDO-Forces calculations with complete geometry optimization have been performed on ac- etaldehyde, vinyl alcohol and acetyl derivatives CH3COX(X=H, F, OH, CN, NH2,NO2,CH3, CF3,OCH3). It was found that acetaldehyde is more stable than vinyl alcohol by 10.451 kcal/mol. Thermodynamically, keto tautomers are more stable than their enol counterparts. This agrees with theoretical calculations. The electron releasing substituents tend to stabilize keto tautomers, while the electron withdrawing substituents tend to destabilize the keto tautomers, relative to the parent. Geometrical parameters, heats of formation, electron densities, Gibbs free energies and orbital ener- gies (HOMO-LUMO) are reported. Key words: Acetaldehyde; Vinyl Alcohol; Keto-Enol Tautomerism; Acetyl Derivative. 1. Introduction 311++G∗∗//MP2(full)/6-31G∗ levels of theory, ac- etaldehyde 1 is found to lie 11.2 and 13.35 kcal/mol Tautomerism refers to the equilibrium between two below vinyl alcohol 2 on the potential energy sur- different structures of the same compound. It is a pro- face, respectively. Recent B3LYP and G2MP2 calcu- totropic rearrangement in which a hydrogen at the α- lations [14] show that acetaldehyde 1 lies 10.4 and position to a carbon-heteroatom double bond migrates 11.1 kcal/mol below vinyl alcohol 2, respectively. -

Determination of Solvent Effects on Ketoðenol Equilibria of 1,3-Dicarbonyl Compounds Using NMR: Revisiting a Classic Physical C
In the Laboratory Determination of Solvent Effects on Keto–Enol Equilibria W of 1,3-Dicarbonyl Compounds Using NMR Revisiting a Classic Physical Chemistry Experiment Gilbert Cook* and Paul M. Feltman Department of Chemistry, Valparaiso University, Valparaiso, IN 46383; *[email protected] “The influence of solvents on chemical equilibria was discovered in 1896, simultaneously with the discovery of keto–enol tautomerism in 1,3-dicarbonyl compounds” (1). The solvents were divided into two groups according to their ability to isomerize compounds. The study of the keto–enol tautomerism of β-diketones and β-ketoesters in a variety of solvents using proton NMR has been utilized as a physical Figure 1. The β-dicarbonyl compounds studied in the experiment. chemistry experiment for many years (2, 3). The first reported use of NMR keto–enol equilibria determination was by Reeves (4). This technique has been described in detail in an experiment by Garland, Nibler, and Shoemaker (2). panded (i) to give an in-depth analysis of factors influencing The most commonly used β-diketone for these experi- solvent effects in tautomeric equilibria and (ii) to illustrate ments is acetylacetone (Scheme I). Use of proton NMR is a the use of molecular modeling in determining the origin of viable method for measuring this equilibrium because the a molecule’s polarity. The experiment’s original benefits of tautomeric keto–enol equilibrium is slow on the NMR time using proton NMR as a noninvasive method of evaluating scale, but enol (2a)–enol (2b) tautomerism is fast on this scale equilibrium are maintained. (5). It has been observed that acyclic β-diketones and β- Experimental Procedure ketoesters follow Meyer’s rule of a shift in the tautomeric equi- librium toward the keto tautomer with increasing solvent Observations of the solvent effects for three other 1,3- polarity (6). -
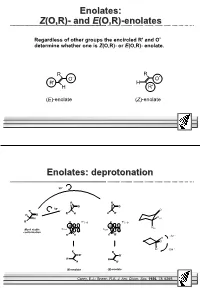
Enolates: Z(O,R)- and E(O,R)-Enolates
Enolates: Z(O,R)- and E(O,R)-enolates Regardless of other groups the encircled R' and O- determine whether one is Z(O,R)- or E(O,R)- enolate. R R O- O- R' H H R' (E)-enolate (Z)-enolate Enolates: deprotonation 90° R R H O H O 30° R O R' H H R' H O H Heq π π R' R *C=O R *C=O H H σ O σ O Hax Most stable C-H C-H conformation R' H H R' -12 ° H O H 104 ° R R - O O- R' H H R' (E)-enolate (Z)-enolate Corey, E.J.; Sneen, R.A. J. Am. Chem. Soc. 1956, 78, 6269. Effect of base on enolisation ! Base must be large and hard. ! Thus functions only as a base and not as a nucleophile. Ph Ph Si Si Si Si - + N N N N O K Li Li M M LDA LiTMP MHMDS ((Me2Ph)2Si)NLi t-BuOK M = Li, Na, K pKa 36 37 26 25 18-20 Selective formation of E/Z- enolates O base TMSCl OTMS OTMS R R R Z(O,R) E(O,R) RbaseZE Et LDA 30 70 (Me3Si)2NLi 70 30 (Et3Si)2NLi 99 1 (Me2PhSi)2NLi >100 <1 cC6H11 LDA 61 39 (Me3Si)2NLi 85 15 (Et3Si)2NLi 94 6 (Me2PhSi)2NLi 99 1 Masamune, S. Aldrichimica Acta 1982, 15, 47. Enolisation: Ireland-mechanism ! According to the Ireland-mechanism an (E)-enolate is formed via a chair form transition state (R = large alkyl group) ! If also R’ is large, a (Z)-enolate is formed! ! Note the actual proton abstractor and the role of the metal! R , LiBr N N Li R H R R R + Li - 78 °C - - O O O H R' O R = Et 50 : 1 "R R = i-Pr 21 : 1 R= t-Bu 1 : >20 Ireland, R.E. -

Bio-Organic Mechanism Game – Simplistic Biochemical Structures and Simplistic Organic Reaction Mechanisms Are Used to Explain Common Biochemical Transformations
1 Bio-Organic Mechanism Game – Simplistic biochemical structures and simplistic organic reaction mechanisms are used to explain common biochemical transformations. Simplified biochemical molecules are presented first. Many biomolecules have a somewhat complex structure that makes it difficult to write out step by step mechanisms. However, if we simplify those structures to the essential parts necessary to explain the mechanistic chemistry of each step, it becomes much easier to consider each step through an important cycle. I have proposed possible simplified structures that are used in the later examples of biochem cycles and problems. The usual strategy in biochem cycles is to just write names, or perhaps, names and a structure. Occasionally a few mechanistic steps are suggested, but almost never is a detailed sequence of mechanistic steps provided. Since it is hard to find such detailed mechanistic steps anywhere (sometimes they are not known) our proposed steps are, of necessity, somewhat speculative. In this book we are not looking for perfection, which is not possible, but for sound organic logic that is consistent with the biochemical examples presented below. There is great satisfaction in blending organic knowledge with real life reactions that help explain how life works. In working through some of the problems, you may develop an alternative mechanism that is just as good, or even better than the one I have proposed. If you do, I hope you will share it with me and if an improved version of this book ever gets written I can include it the next edition (and give you credit). It is almost certain that I have made some errors and I would appreciate it if you would let me know about them. -
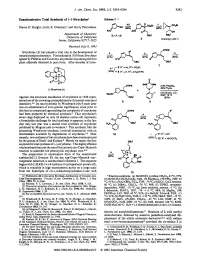
Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1
J. Am. Chem. SOC.1993,115, 9293-9294 9293 Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1 Steven D. Knight, Larry E. Overman,' and Garry Pairaudeau Department of Chemistry University of California Imine, California 9271 7-2025 Received July 8, 1993 OTlPS Strychnine (1) has played a vital role in the development of d h,i natural productschemistry. First isolated in 1818 fromstrychnos ignatii by Pelletier and Caventou, strychnine was among the first plant alkaloids obtained in pure form. After decades of inves- OBU' (-)-Strychnine(1) tigation, the structural elucidation of strychnine in 1946 repre- sented one of the crowning accomplishments of classical structural chemistry.394 Its total synthesis by Woodward only 8 years later was an achievement of even greater significance, since prior to this feat no compound approaching the complexity of strychnine ,oms had been prepared by chemical synthesis.* That strychnine's P' seven rings displayed on only 24 skeletal atoms still represents a formidable challenge for total synthesis is apparent in the fact that only last year was a second total synthesis of strychnine published by Magnus and co-workers.6 This synthesis, like the pioneering Woodward synthesis, involved intersection with an intermediate available by degradation of stry~hnine.~.~Most recently, two syntheses of (*)-strychnine have been communicated by the groups of Stork' and Kuehne.* Herein we report the first asymmetric total synthesis of (-)-strychnine. This highly efficient total synthesis features the use of the cationic aza-Cope-Mannich reaction to assemble the pentacyclic strychnan c~re.~J~ The preparation in enantiopure form of the unsaturated azabicyclo[3.2. -

Fischer Indole Synthesis Applied to the Total Synthesis of Natural Products Cite This: RSC Adv.,2017,7,52852 Majid M
RSC Advances REVIEW View Article Online View Journal | View Issue Fischer indole synthesis applied to the total synthesis of natural products Cite this: RSC Adv.,2017,7,52852 Majid M. Heravi, * Sahar Rohani, Vahideh Zadsirjan* and Nazli Zahedi One of the oldest and most useful reactions in organic chemistry is the Fischer indole synthesis (FIS). It is known to have a wide variety of applications including the synthesis of indole rings, often present as the Received 27th September 2017 framework in the total synthesis of natural products, particularly those found in the realm of alkaloids, Accepted 3rd November 2017 which comprise a ring system known as an indole alkaloid. In this review, we are trying to emphasize the DOI: 10.1039/c7ra10716a applications of FIS as an old reaction, which is currently applied to the total synthesis of biologically rsc.li/rsc-advances active natural products and some other complex targets. 1. Introduction prescribed drugs or medications.1 Remarkably, the name indole is a combination of the words indigo and oleum because Creative Commons Attribution 3.0 Unported Licence. Nowadays, there has been increasing attention on the total initially, indole was prepared and identied from the reaction synthesis of bioactive natural products and their synthetic of the indigo dye with oleum.2 As a matter of fact, one of the analogues in the arena of organic chemistry. Novel synthetic most plentiful heterocyclic systems found in nature is indole. It approaches and strategies are available that permit formation is a vital functional nucleus in the structures of different dyes, of novel complex molecules or already structurally known fragrances, pharmaceuticals and agricultural chemicals.3,4 naturally occurring compounds which can be used as Indole ring moieties became important structural components in diverse natural pharmaceutical agents, hence their synthesis and functionalization is a key eld in heterocyclic chemistry, Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran. -

Summary Sheet 2: Enols and Enolates
Summary Sheet 2: Enols and Enolates Name Structure Example pKA* of H Structural Features of the Carbonyl Group: Effects on acidity of alkyl groups Effect on reactivity of alkenes: 2 O O •Carbon, oxygen: sp hybridized Likewise, the presence of a carbonyl group activates alkenes toward The carbonyl is an electron withdrawing system with aldehyde 17 •O–C–C bond angle ~120 ° π nucleophilic attack: low-lying π* orbitals. It stabilizes adjacent negative charge. R H H3C H •C=O bond strongly polarized O – δ toward oxygen. M H3C CH3 M H H H O O •Carbonyl carbon is partially positive H2C CH3 LiNEt2 CH X CH3 δ+ 2 does not form ketone 20 therefore electrophilic! Ethane R R' H C Me Conjugate base NEt2 3 •Lone pairs render oxygen weakly pKa = ~50 H H NEt2 nucleophilic (will react with strong acid) highly unstable O O OMe OMe ester 25 O O O Li O O R OR H C OMe Key Concept: Tautomerism CH3 CH3 3 H t-Bu H t-Bu t-Bu M O LiNEt2 O H X O O O O OH Methyl propionate Conjugate base (enolate) lactone pKa = ~25 ~1025 more acidic just by H H Et2N Et2N O H C O H H (cyclic 2 C R R replacing H with a carbonyl! ( ) H enolate: stabilized! forms readily! ester) n 2 H Why the huge difference in acidity? The lone pair is keto form enol form stabilized by donation into the carbonyl π system. The reactivity of the alkene toward nucleophilic attack is directly related O to the stability of the enolate that forms - O Tautomerism: a form of isomerism where OMe OMe amide 30 a keto converts to an enol through the movement of O O R NR CH3 CH3 O O 2 H2C NMe2 a proton and shifting of bonding electrons O O O O M M NH2 = 1° amide For acetone (R=CH ) the keto:enol ratio is ~6600:1 at 23 > > > > 3 NR2 OR Me CF RO OR NHR = 2 ° amide °C. -

Enols and Enolates 22.1 Introduction to Alpha Carbon Chemistry
4/25/2012 22.1 Introduction to Alpha Carbon 22.1 Introduction to Alpha Carbon Chemistry –Enols and Enolates Chemistry –Enols and Enolates • For carbonyl compounds, Greek letters are often used • The reactions we will explore proceed though either an to describe the proximity of atoms to the carbonyl enol or an enolate intermediate. center. • This chapter will primarily explore reactions that take place at the alpha carbon. Copyright 2012 John Wiley & Sons, Inc. 22-1 Klein, Organic Chemistry 1e Copyright 2012 John Wiley & Sons, Inc. 22-2 Klein, Organic Chemistry 1e 22.1 Introduction to Alpha Carbon 22.1 Introduction to Alpha Carbon Chemistry –Enols and Enolates Chemistry –Enols and Enolates • Trace amounts of acid or base catalyst provide • In rare cases such as the example below, the enol form equilibriums in which both the enol and keto forms are is favored in equilibrium. present. • Give two reasons to explain WHY the enol is favored. • How is equilibrium different from resonance? • At equilibrium, > 99% of the molecules exist in the keto • The solvent can affect the exact percentages. form. WHY? Copyright 2012 John Wiley & Sons, Inc. 22-3 Klein, Organic Chemistry 1e Copyright 2012 John Wiley & Sons, Inc. 22-4 Klein, Organic Chemistry 1e 22.1 Introduction to Alpha Carbon 22.1 Introduction to Alpha Carbon Chemistry –Enols and Enolates Chemistry –Enols and Enolates • Phenol is an example where the enol is vastly favored • The mechanism for the tautomerization depends on over the keto at equilibrium. WHY? whether it is acid catalyzed or base catalyzed. Copyright 2012 John Wiley & Sons, Inc.