Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -
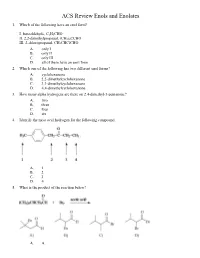
Ochem ACS Review 18 Enols and Enolates
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Abstract Multicomponent Reactions of Salicylaldehyde, Cyclic Ketones, and Arylamines Through Cooperative Enamine-Metal Lewis
ABSTRACT MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS by Ryan Gregory Sarkisian Multicomponent reactions (MCRs) are the most atom economic, highly selective, and convergent type of reaction. This allows for a reaction to have a wide scope and allows for maximization of the complexity of a product. Catalyzing these MCRs with asymmetric catalysis is a novel way to introduce stereocontrol into highly complex molecules with various functional groups. Asymmetric catalysis is considered the most efficient method for constructing highly functionalized optically active stereopure compounds. There are three pillars of asymmetric catalysis: biocatalysis, transition metal catalysis, and organocatalysis. This research focuses on two of these pillars, transition metal catalysis and organocatalysis, working cooperatively to catalyze this MCR. The focus is to educate or refresh the audience on the basic topics that make up the complexity of the MCRs being catalyzed by cooperative asymmetric catalysis. Ultimately to explore the cooperative catalysts used to synthesize both the racemic and asymmetric three fused ring products (9-((4-methoxyphenyl)amino)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-4a-ol). MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS A THESIS Submitted to the Faculty of Miami University in partial fulfillment of the requirements for the degree of Master of Science Department of -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -
![Synthesis of Densely Substituted Pyridine Derivatives from Nitriles by a Non-Classical [4+2] Cycloaddition/1,5-Hydrogen Shift Strategy](https://docslib.b-cdn.net/cover/3428/synthesis-of-densely-substituted-pyridine-derivatives-from-nitriles-by-a-non-classical-4-2-cycloaddition-1-5-hydrogen-shift-strategy-483428.webp)
Synthesis of Densely Substituted Pyridine Derivatives from Nitriles by a Non-Classical [4+2] Cycloaddition/1,5-Hydrogen Shift Strategy
Synthesis of densely substituted pyridine derivatives from nitriles by a non-classical [4+2] cycloaddition/1,5-hydrogen shift strategy Wanqing Wu ( [email protected] ) South China University of Technology https://orcid.org/0000-0001-5151-7788 Dandan He South China University of Technology Kanghui Duan South China University of Technology Yang Zhou South China University of Technology Meng Li South China University of Technology Huanfeng Jiang South China University of Technology Article Keywords: pyridine derivatives, organic synthesis, medicinal chemistry. Posted Date: March 3rd, 2021 DOI: https://doi.org/10.21203/rs.3.rs-258126/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract A novel strategy has been established to assemble an array of densely substituted pyridine derivatives from nitriles and o-substituted aryl alkynes or 1-methyl-1,3-enynes via a non-classical [4 + 2] cycloaddition along with 1,5-hydrogen shift process. The well-balanced anities of two different alkali metal salts enable the C(sp3)-H bond activation as well as the excellent chemo- and regioselectivities. This protocol offers a new guide to construct pyridine frameworks from nitriles with sp3-carbon pronucleophiles, and shows potential applications in organic synthesis and medicinal chemistry. Introduction Compounds containing pyridine core structures, not only widely exist in natural products, pharmaceuticals, and functional materials,1–7 but also serve as useful and valuable building blocks for metal ligands.8,9 For instance, pyridine derivative Actos I10 is a famous drug for the treatment of type 2 diabetes; Bi-(or tri-)pyridines II11–15 are often used as ligands in metal-catalyzed reactions; Kv1.5 antagonist III16 and alkaloid papaverine IV17 are two representative isoquinolines as a promising atrial- selective agent and a smooth muscle relaxant, respectively (Fig. -

TR-470: Pyridine (CASRN 110-86-1) in F344/N Rats, Wistar Rats, And
NTP TECHNICAL REPORT ON THE TOXICOLOGY AND CARCINOGENESIS STUDIES OF PYRIDINE (CAS NO. 110-86-1) IN F344/N RATS, WISTAR RATS, AND B6C3F1 MICE (DRINKING WATER STUDIES) NATIONAL TOXICOLOGY PROGRAM P.O. Box 12233 Research Triangle Park, NC 27709 March 2000 NTP TR 470 NIH Publication No. 00-3960 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service National Institutes of Health FOREWORD The National Toxicology Program (NTP) is made up of four charter agencies of the U.S. Department of Health and Human Services (DHHS): the National Cancer Institute (NCI), National Institutes of Health; the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health; the National Center for Toxicological Research (NCTR), Food and Drug Administration; and the National Institute for Occupational Safety and Health (NIOSH), Centers for Disease Control and Prevention. In July 1981, the Carcinogenesis Bioassay Testing Program, NCI, was transferred to the NIEHS. The NTP coordinates the relevant programs, staff, and resources from these Public Health Service agencies relating to basic and applied research and to biological assay development and validation. The NTP develops, evaluates, and disseminates scientific information about potentially toxic and hazardous chemicals. This knowledge is used for protecting the health of the American people and for the primary prevention of disease. The studies described in this Technical Report were performed under the direction of the NIEHS and were conducted in compliance with NTP laboratory health and safety requirements and must meet or exceed all applicable federal, state, and local health and safety regulations. Animal care and use were in accordance with the Public Health Service Policy on Humane Care and Use of Animals. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C.