Abstract Multicomponent Reactions of Salicylaldehyde, Cyclic Ketones, and Arylamines Through Cooperative Enamine-Metal Lewis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

Comparative Total Syntheses of Strychnine
Comparative Total Syntheses of Strychnine N C D MacMillan Group Meeting N H O H A H B E Nathan Jui H N N H H F G July 22, 2009 O O O References: Pre-Volhardt: Bonjoch Chem. Rev. 2000, 3455. Woodward Tetrahedron, 1963, 247. Volhardt J. Am. Chem. Soc. 2001, 9324. Magnus J. Am. Chem. Soc. 1993, 8116. Martin J. Am. Chem. Soc. 2001, 8003. Overman J. Am. Chem. Soc. 1995, 5776. Bodwell Angew. Chem. Int. Ed. 2002, 3261. Kuehne J. Org. Chem. 1993, 7490. Mori J. Am. Chem. Soc. 2003, 9801. Kuehne J. Org. Chem. 1998, 9427. Shibasaki Tetrahedron 2004, 9569. Rawal J. Org. Chem. 1994, 2685. Fukuyama J. Am. Chem. Soc. 2004, 10246. Bosch, Bonjoch Chem. Eur. J. 2000, 655. Padwa Org. Lett. 2007, 279. History and Structure of (!)-Strychnine ! Isolated in pure form in 1818 (Pelletier and Caventou) ! Structural Determination in 1947 (Robinson and Leuchs) ! 24 skeletal atoms (C21H22N2O2) ! Isolated in pure form in 1818 (Pelletier and Caventou) N ! Over !25 07 -pruinbglisc,a 6ti-osntesr epoecrteaninteinrgs to structure ! Structural Determination in 1947 (Robinson and Leuchs) ! Notor!io u Ss ptoirxoicne (nletethr a(lC d-o7s) e ~10-50 mg / adult) N H H ! Over 250 publications pertaining to structure O O ! Comm!o nClyD uEs eridn gr osdyesntet mpoison ! Notorious poison (lethal dose ~10-50 mg / adult) ! Hydroxyethylidine Strychnos nux vomica ! $20.20 / 10 g (Aldrich), ~1.5 wt% (seeds), ~1% (blossoms) "For it's molecular size it is the most complex substance known." -Robert Robinson History and Structure of (!)-Strychnine ! 24 skeletal atoms (C21H22N2O2) N ! 7-rings, 6-stereocenters ! Spirocenter (C-7) N H H O O ! CDE ring system ! Hydroxyethylidine "For it's molecular size it is the most complex substance known." -Robert Robinson "If we can't make strychnine, we'll take strychnine" -R. -
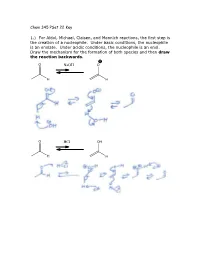
Chem 345 Pset 22 Key 1.) for Aldol, Michael, Claisen, and Mannich
Chem 345 PSet 22 Key 1.) For Aldol, Michael, Claisen, and Mannich reactions, the first step is the creation of a nucleophile. Under basic conditions, the nucleophile is an enolate. Under acidic conditions, the nucleophile is an enol. Draw the mechanism for the formation of both species and then draw the reaction backwards. O NaOH O H H O HCl OH H H 2.) In the case of a Mannich reaction, the electrophile (an imine) must also be made. Draw the mechanism for the forward and backward reaction: O MeNH2 N H H cat. AcOH H H 3.) When a secondary amine is used and the carbonyl has an enolizable proton, an enamine can be formed. Are enamines nucleophilic or electrophilic? Nucleophilic H O N N H2O H cat. AcOH H enamine N N H H enamine 4.) The following reaction is the Lobry-de-Bruyn-von-Ekenstein reaction. Don’t worry about the name. I only recently found out that that was the official name for the rearrangement leading from glucose to fructose or mannose and back again. I was taught it under a different name: the Carbohydrate Game. Under acidic or basic conditions, it is possible that the following sugars (glucose, fructose, and mannose) are in equilibrium with each other. Draw the acid catalyzed version and the base catalyzed version. (Hint: this is just a repeat of question 1.) O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose The sugars are drawn in a Fisher projection (or what I think of is a dead cat projection). -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics -

Mannich Reaction
1. Introduction 1.1- Mannich Reaction The Mannich reaction is three component condensation in which a compound containing an active hydrogen atom is allowed to react with formaldehyde and an NH-amine derivative . Secondary amines rather than primary amines and ammonia are employed , the resulting product (Mannich Base ) is an amine compound having the N atom linked to the R substrate through a methylene group 1,2. The aminoalkylation of CH-acidic compounds was described by several authors as early as the 19th. century. However, it was Carl Mannich who was the first to recognize the enormous significance of this reaction , and it was he who extended the chemistry into a broad based synthetic methodology through systematic research. Since then this reaction that now carries his name has developed into one of the most important C-C bond-forming reactions in organic chemistry3,4. The Mannich reaction is a classical method for the preparation of β -aminoketones and aldehydes (Mannich bases) and, as such, is one of the most important basic reaction types in organic chemistry. It is the key step in the synthesis of numerous pharmaceuticals and natural products3,4. 1 The Mannich reaction can be presented by the following equation: The essential feature of the reaction is the replacement of the active hydrogen atom by an aminomethyl or substituted aminomethyl group. The symbolizes the active hydrogen component which includes ketones, aldehydes, acids, esters, phenols, acetylenes, picolines, nitroalkanes and quinolines1,2. 1.2- Mechanism of the Mannich reaction The mechanism of the Mannich reaction has been well investigated, the condensation reaction occurs in two steps: first, the amine reacts with formaldehyde to give condensation product (5 ), (6), ( 7) (step I) which then attacks the substrate R-H (step II) .The reaction does not normally follow the other possible route (step III and IV) ; however, some successful reaction between hydroxymethyl derivatives (8) and alkylamines to give Mannich bases (9) should be mentioned . -
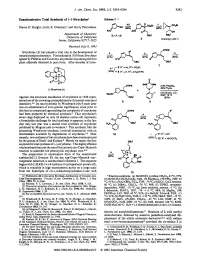
Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1
J. Am. Chem. SOC.1993,115, 9293-9294 9293 Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1 Steven D. Knight, Larry E. Overman,' and Garry Pairaudeau Department of Chemistry University of California Imine, California 9271 7-2025 Received July 8, 1993 OTlPS Strychnine (1) has played a vital role in the development of d h,i natural productschemistry. First isolated in 1818 fromstrychnos ignatii by Pelletier and Caventou, strychnine was among the first plant alkaloids obtained in pure form. After decades of inves- OBU' (-)-Strychnine(1) tigation, the structural elucidation of strychnine in 1946 repre- sented one of the crowning accomplishments of classical structural chemistry.394 Its total synthesis by Woodward only 8 years later was an achievement of even greater significance, since prior to this feat no compound approaching the complexity of strychnine ,oms had been prepared by chemical synthesis.* That strychnine's P' seven rings displayed on only 24 skeletal atoms still represents a formidable challenge for total synthesis is apparent in the fact that only last year was a second total synthesis of strychnine published by Magnus and co-workers.6 This synthesis, like the pioneering Woodward synthesis, involved intersection with an intermediate available by degradation of stry~hnine.~.~Most recently, two syntheses of (*)-strychnine have been communicated by the groups of Stork' and Kuehne.* Herein we report the first asymmetric total synthesis of (-)-strychnine. This highly efficient total synthesis features the use of the cationic aza-Cope-Mannich reaction to assemble the pentacyclic strychnan c~re.~J~ The preparation in enantiopure form of the unsaturated azabicyclo[3.2. -

Asymmetric Catalysis in Aqueous Media*
Pure Appl. Chem., Vol. 79, No. 2, pp. 235–245, 2007. doi:10.1351/pac200779020235 © 2007 IUPAC Asymmetric catalysis in aqueous media* Shu– Kobayashi Graduate School of Pharmaceutical Sciences, The University of Tokyo, The HFRE Division, ERATO, Japan Science and Technology Agency (JST), Hongo, Bunkyo-ku, Tokyo 113-0033, Japan Abstract: Lewis acid catalysis has attracted much attention in organic synthesis because of unique reactivity and selectivity attained under mild conditions. Although various kinds of Lewis acids have been developed and applied in industry, these Lewis acids must be gener- ally used under strictly anhydrous conditions. The presence of even a small amount of water handles the reactions owing to preferential reactions of the Lewis acids with water rather than the substrates. In contrast, rare earth and other metal complexes have been found to be water- compatible. Several catalytic asymmetric reactions in aqueous media, including hydroxy- methylation of silicon enolates with an aqueous solution of formaldehyde in the presence of Sc(OTf)3–chiral bipyridine ligand or Bi(OTf)3–chiral bipyridine ligand, Sc- or Bi-catalyzed asymmetric meso-epoxide ring-opening reactions with amines, and asymmetric Mannich- type reactions of silicon enolates with N-acylhydrazones in the presence of a chiral Zn cata- lyst have been developed. Water plays key roles in these asymmetric reactions. Keywords: reactions in aqueous media; Lewis acids; catalytic asymmetric reactions; hydroxymethylation of silicon enolates; Sc(OTf)3–chiral bipyridine ligand. INTRODUCTION Organic reactions in aqueous media are of current interest owing to the key role played by water as a solvent for green chemistry [1]. -

Asymmetric Synthesis of C–F Quaternary Α-Fluoro-Β-Amino-Indolin
RSC Advances View Article Online PAPER View Journal | View Issue Asymmetric synthesis of C–F quaternary a-fluoro- b-amino-indolin-2-ones via Mannich addition Cite this: RSC Adv.,2017,7,5679 reactions; facets of reactivity, structural generality and stereochemical outcome† Chen Xie,a Wanxing Sha,a Yi Zhu,a Jianlin Han,*a Vadim A. Soloshonokbc and Yi Pana Reported herein is a new approach for the preparation of enantiomerically pure a-fluoro-b-amino-indolin- Received 3rd December 2016 2-ones possessing tetrasubstituted fluorinated stereogenic centers. This method includes the Accepted 27th December 2016 detrifluoroacetylative in situ generation of tertiary enolates followed by Mannich reaction with (Ss)- DOI: 10.1039/c6ra27710a sulfinylimines. The operationally convenient conditions coupled with perfect diastereoselectivity and www.rsc.org/advances functional substituent compatibility bode well for widespread application of this new synthetic method. Creative Commons Attribution 3.0 Unported Licence. Fluoro-organic chemistry plays an essential role in furthering solutions. Over the last several years, our group has contributed numerous technological advancements, most notably in the to this research area by the development of cyclic enolates 2 energy,1 food2 and healthcare industries.3 Consequently, the (eqn (2)) allowing preparation of structurally complex products current interest in the synthesis of uorine-containing possessing tetrasubstituted uorinated stereogenic centers.13 compounds is at an all-time high, addressing the emerging The -

The Robinson-Mannich Base Synthesis Applied to Substituted
THE ROBINSON- MANNICH BASE SYNTHESIS APPLIED TO SUBSTITUTED OETOACETIC ESTERS by RICHARD FRANCIS LAPOBli: A THESIS subln1tted to OREGON 6TA7!E COI..t..iX}li: in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY June 1954 IXllffiDT Redacted for Privacy t & errur of h;lar Redacted for Privacy 6r1*m of Bq:stmt Redacted for Privacy chrtflra sqr fiel Sreffir ffit* Redacted for Privacy IH rf ffi&rr*r l**rrfi Lt tlrmlr lr firr $rpri ry lu.jltr Frlrtrt'rlrrr TABLE OF CONTENTS Page Introduction. • • . • .. • • • •· • • .. 1 B1stor1caJ.. • • • • • • • • • • • 4 Discussion of Results .. .• • .. • • • • • • • • • • 9 Experimental. • • • • • • • ·• • .. 32 Summary • • • . .. .. ,,.. • .. • • • • • . • • • • 42 Bibliography. • .. .. • .. .. • • • • • • • • • " LISf OF FIGURES Figure 1. • • • • • • ., • • . 3 Figure 2. • • • .. .. • • .. .. • • • • .. • • • .. • 6 F1:gure ... 3. •· " • • • • • • • 29 Figure 4. • • • • ·• .. • . ·• • • • • • • 30 F1gure 5. • • • • ,. • • • '1 ACKNOWLEDGMENT The author \'T1sbes to express his thanks to the E. I . duPont de Nemours and OompallJ (Inc. ) for assistance during the 1952-1953 y-ear in the form of a pre-doctoral fellowship. Gx-atef'ul thanks are also e;xtended to DP. Albert V.. Logan :for his guidance and help while this ~1ork was 1n progress and to his colleague, Dr. E. N. Marvell. :for his helpful suggestions. This work was supported. 1n part b7 the U. s. Atomic Energy Commission under contract with Oregon State College. Ded1cated to my wire. Genie THE ROBINSON- NICH BASE SYNTHESIS APPLIED TO SUBSTITUTED ACETOACETIC ESTERS INTRODUCTION The purpose of this investigation was to determine the feasibility of synthesizing certain substituted naphthalenes containing a cl4 atom placed in a known posi t ion in one of the rings. A contemplated study of the mode, of reaction of the Jacobsen rearrangement made it desirable to know whether or not these compounds m1g}lt be made conveniently, 1n a high state of purity and in good yield. -

One-Pot Enol Silane Formation-Mukaiyama–Mannich
CORE Metadata, citation and similar papers at core.ac.uk Provided by University of Richmond University of Richmond UR Scholarship Repository Chemistry Faculty Publications Chemistry 9-2013 One-Pot Enol Silane Formation- Mukaiyama–Mannich Addition of Ketones, Amides, and Thioesters to Nitrones in the Presence of Trialkylsilyl Trifluoromethanesulfonates C. Wade Downey University of Richmond, [email protected] Carolyn M. Dombrowski Erin N. Maxwell Chelsea L. Safran Odamea A. Akomah Follow this and additional works at: http://scholarship.richmond.edu/chemistry-faculty- publications Part of the Organic Chemistry Commons This is a pre-publication author manuscript of the final, published article. Recommended Citation Downey, C. Wade; Dombrowski, Carolyn M.; Maxwell, Erin N.; Safran, Chelsea L.; and Akomah, Odamea A., "One-Pot Enol Silane Formation-Mukaiyama–Mannich Addition of Ketones, Amides, and Thioesters to Nitrones in the Presence of Trialkylsilyl Trifluoromethanesulfonates" (2013). Chemistry Faculty Publications. 10. http://scholarship.richmond.edu/chemistry-faculty-publications/10 This Post-print Article is brought to you for free and open access by the Chemistry at UR Scholarship Repository. It has been accepted for inclusion in Chemistry Faculty Publications by an authorized administrator of UR Scholarship Repository. For more information, please contact [email protected]. FULL PAPER DOI: 10.1002/ejoc.200 ((will be filled in by the editorial staff)) One-Pot Enol Silane Formation-Mukaiyama–Mannich Addition of Ketones, Amides, and Thioesters to Nitrones in the Presence of Trialkylsilyl Trifluoromethanesulfonates C. Wade Downey,* [a] Carolyn M. Dombrowski, [a] Erin N. Maxwell, [a] Chelsea L. Safran, [a] and Odamea A. Akomah [a] Keywords: Nitrones / Mukaiyama addition / Mannich addition / Silyl triflates / hydroxylamines Ketones, amides, and thioesters form enol silanes and add to N- ((Abstract Text----Continued)) phenylnitrones in one pot in the presence of trimethylsilyl trifluoromethanesulfonate and trialkylamine. -

Development of the Conjugate Addition/Nitro-Mannich Reaction
Development of the conjugate addition/Nitro-Mannich reaction A Thesis presented by Andreas Kalogirou In Part Fulfilment of the requirement for the degree of Doctor of Philosophy University College London January 2013 UCL Chemistry Department 20 Gordon Street London WC1H 0AJ Andreas Kalogirou I, Andreas Kalogirou confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed………………………………………………………… Date…………………………………………………………… University College London 2 Andreas Kalogirou Abstract This thesis describes the development of the conjugate addition/nitro-Mannich reaction and its use in the synthesis of useful molecules like pyrrolidin-2-ones and piperazin-2-ones. The introductory chapter of this thesis outlines the literature related to the nitro-Mannich reaction, describing the different existing methodologies for performing the reaction in diastereo- and enantioselective manner. The synthetic applications of the reaction are also described, especially its uses in the synthesis of biologically active natural products. Moreover, the syntheses and uses of two classes of compounds, pyrrolidin-2-ones and piperazin-2-ones are briefly discussed. The results and discussion chapter starts by presenting the use of a one-pot conjugate addition/nitro-Mannich/lactamisation reaction in the synthesis of densely functionalised pyrrolidin-2-ones. The development of an asymmetric protocol, as well as some functionalisations of the pyrrolidinone core are also described. Our efforts to synthesise human neutrophil elastase inhibitor GW311616A using the developed methodology are then detailed. The next part of the results and discussion chapter portrays the development of a conjugate addition/nitro-Mannich reaction of non-zinc nucleophiles to ethyl 3- nitroacrylate and β-nitrostyrene.