Asymmetric Catalysis in Aqueous Media*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Advances in Titanium Radical Redox Catalysis
JOCSynopsis Cite This: J. Org. Chem. 2019, 84, 14369−14380 pubs.acs.org/joc Recent Advances in Titanium Radical Redox Catalysis Terry McCallum, Xiangyu Wu, and Song Lin* Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States ABSTRACT: New catalytic strategies that leverage single-electron redox events have provided chemists with useful tools for solving synthetic problems. In this context, Ti offers opportunities that are complementary to late transition metals for reaction discovery. Following foundational work on epoxide reductive functionalization, recent methodological advances have significantly expanded the repertoire of Ti radical chemistry. This Synopsis summarizes recent developments in the burgeoning area of Ti radical catalysis with a focus on innovative catalytic strategies such as radical redox-relay and dual catalysis. 1. INTRODUCTION a green chemistry perspective, the abundance and low toxicity of Ti make its complexes highly attractive as reagents and Radical-based chemistry has long been a cornerstone of 5 1 catalysts in organic synthesis. synthetic organic chemistry. The high reactivity of organic IV/III radicals has made possible myriad new reactions that cannot be A classic example of Ti -mediated reactivity is the reductive ring opening of epoxides. This process preferentially readily achieved using two-electron chemistry. However, the − high reactivity of organic radicals is a double-edged sword, as cleaves and functionalizes the more substituted C O bond, the selectivity of these fleeting intermediates can be difficult to providing complementary regioselectivity to Lewis acid control in the presence of multiple chemotypes. In addition, promoted epoxide reactions. The synthetic value of Ti redox catalysis has been highlighted by their many uses in total catalyst-controlled regio- and stereoselective reactions involv- 6−10 ing free-radical intermediates remain limited,2 and the synthesis (Scheme 1). -

Abstract Multicomponent Reactions of Salicylaldehyde, Cyclic Ketones, and Arylamines Through Cooperative Enamine-Metal Lewis
ABSTRACT MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS by Ryan Gregory Sarkisian Multicomponent reactions (MCRs) are the most atom economic, highly selective, and convergent type of reaction. This allows for a reaction to have a wide scope and allows for maximization of the complexity of a product. Catalyzing these MCRs with asymmetric catalysis is a novel way to introduce stereocontrol into highly complex molecules with various functional groups. Asymmetric catalysis is considered the most efficient method for constructing highly functionalized optically active stereopure compounds. There are three pillars of asymmetric catalysis: biocatalysis, transition metal catalysis, and organocatalysis. This research focuses on two of these pillars, transition metal catalysis and organocatalysis, working cooperatively to catalyze this MCR. The focus is to educate or refresh the audience on the basic topics that make up the complexity of the MCRs being catalyzed by cooperative asymmetric catalysis. Ultimately to explore the cooperative catalysts used to synthesize both the racemic and asymmetric three fused ring products (9-((4-methoxyphenyl)amino)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-4a-ol). MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS A THESIS Submitted to the Faculty of Miami University in partial fulfillment of the requirements for the degree of Master of Science Department of -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Asymmetric Lewis Acid Catalysis Directed by Octahedral Rhodium Centrochirality† Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Asymmetric Lewis acid catalysis directed by octahedral rhodium centrochirality† Cite this: Chem. Sci.,2015,6,1094 Chuanyong Wang,‡a Liang-An Chen,‡b Haohua Huo,a Xiaodong Shen,a Klaus Harms,a Lei Gongb and Eric Meggers*ab A rhodium-based asymmetric catalyst is introduced which derives its optical activity from octahedral centrochirality. Besides providing the exclusive source of chirality, the rhodium center serves as a Lewis acid by activating 2-acyl imidazoles through two point binding and enabling a very effective asymmetric induction mediated by the propeller-like C2-symmetrical ligand sphere. Applications to asymmetric Michael additions (electrophile activation) as well as asymmetric a-aminations (nucleophile activation) Received 9th October 2014 are disclosed, for which the rhodium catalyst is found to be overall superior to its iridium congener. Due Accepted 7th November 2014 to its straightforward proline-mediated synthesis, high catalytic activity (catalyst loadings down to 0.1 DOI: 10.1039/c4sc03101f mol%), and tolerance towards moisture and air, this novel class of chiral-at-rhodium catalysts will likely www.rsc.org/chemicalscience to become of widespread use as chiral Lewis acid catalysts for a large variety of asymmetric transformations. Creative Commons Attribution 3.0 Unported Licence. Lewis acids are capable of activating a large variety of carbon- now wish to report for the rst time that rhodium can also serve heteroatom and carbon–carbon bond forming reactions and as the combined source of centrochirality and Lewis acidity in chiral Lewis acids have therefore become indispensable tools substitutionally labile octahedral metal complexes. -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

Physical Organic Chemistry
CHM 8304 Physical Organic Chemistry Catalysis Outline: General principles of catalysis • see section 9.1 of A&D – principles of catalysis – differential bonding 2 Catalysis 1 CHM 8304 General principles • a catalyst accelerates a reaction without being consumed • the rate of catalysis is given by the turnover number • a reaction may alternatively be “promoted” (accelerated, rather than catalysed) by an additive that is consumed • a heterogeneous catalyst is not dissolved in solution; catalysis typically takes place on its surface • a homogeneous catalyst is dissolved in solution, where catalysis takes place • all catalysis is due to a decrease in the activation barrier, ΔG‡ 3 Catalysts • efficient at low concentrations -5 -4 -5 – e.g. [Enz]cell << 10 M; [Substrates]cell < 10 - 10 M • not consumed during the reaction – e.g. each enzyme molecule can catalyse the transformation of 20 - 36 x 106 molecules of substrate per minute • do not affect the equilibrium of reversible chemical reactions – only accelerate the rate of approach to equilibrium end point • most chemical catalysts operate in extreme reaction conditions while enzymes generally operate under mild conditions (10° - 50 °C, neutral pH) • enzymes are specific to a reaction and to substrates; chemical catalysts are far less selective 4 Catalysis 2 CHM 8304 Catalysis and free energy • catalysis accelerates a reaction by stabilising a TS relative to the ground state ‡ – free energy of activation, ΔG , decreases – rate constant, k, increases • catalysis does not affect the end point -

Research in the Department of Homogeneous Catalysis
Homogeneous Catalysis – Overview Research in the Department of Homogeneous Catalysis Researchers in our department continue focusing on the development of new catalysis concepts within the areas of organocatalysis and transition metal catalysis. We explore new catalysts, expand the substrate scope of certain catalytic reactions, apply asymmetric catalysis in natural product and pharmaceutical synthesis, and study mechanisms of homogenous catalytic reactions (B. List, K.-R. Pörschke, M. Klußmann, N. Maulide). Since leadership of the department of homogenous catalysis was taken over by Professor Benjamin List in 2005, it has grown significantly from ca. 15 members to currently > 50 members, including the groups of Professor K.-R. Pörschke, who has been a group leader at the institute since two decades now, Dr. M. Klußmann (group leader since 2007), and now also of Dr. N. Maulide, who has joined the department in 2009. The group of Professor List primarily advances enantioselective organocatalysis as a fundamental approach complementing the already more advanced fields of biocatalysis and transition metal catalysis. The List group has a profound interest in developing “new reactions”, designs and identifies new principles for the development of organocatalysts, expands the scope of already developed catalysts such as proline, uses organocatalysis in the synthesis of natural products and pharmaceuticals, and also investigates the mechanism by which organocatalysts activate their substrates. Furthermore, in 2005 the group has first conceptualized and then significantly advanced another approach to asymmetric catalysis, asymmetric counteranion directed catalysis (ACDC). Initially, merely an idea, this approach has progressed within the department but now also at other institutions around the globe, into a truly general strategy for asymmetric synthesis and has found utility in organocatalysis but also in transition metal catalysis and Lewis acid catalysis. -

Chapter 8. Chiral Catalysts José M
Chapter 8. Chiral Catalysts José M. Fraile, José I. García, José A. Mayoral 1. The Origin of Enantioselectivity in Catalytic Processes: the Nanoscale of Enantioselective Catalysis. Enantiomerically pure compounds are extremely important in fields such as medicine and pharmacy, nutrition, or materials with optical properties. Among the different methods to obtain enantiomerically pure compounds, asymmetric catalysis1 is probably the most interesting and challenging, in fact one single molecule of chiral catalyst can transfer its chiral information to thousands or even millions of new chiral molecules. Enantioselective reactions are the result of the competition between different possible diastereomeric reaction pathways, through diastereomeric transition states, when the prochiral substrate complexed to the chiral catalyst reacts with the corresponding reagent. The efficiency of the chirality transfer, measured as enantiomeric excess [% ee = (R−S)/(R+S) × 100], depends on electronic and steric factors in a very subtle form. A simple calculation shows that differences in energy of only 2 kcal/mol between these transition states are enough to obtain more than 90% ee, and small changes in any of the participants in the catalytic process can modify significantly this difference in energy. Those modifications may occur in the near environment of the catalytic centre, at less than 1 nm scale, but also at longer distances in the catalyst, substrate, reagent, solvent, or support in the case of immobilized catalysts. This is the reason because asymmetric -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

Comparative Total Syntheses of Strychnine
Comparative Total Syntheses of Strychnine N C D MacMillan Group Meeting N H O H A H B E Nathan Jui H N N H H F G July 22, 2009 O O O References: Pre-Volhardt: Bonjoch Chem. Rev. 2000, 3455. Woodward Tetrahedron, 1963, 247. Volhardt J. Am. Chem. Soc. 2001, 9324. Magnus J. Am. Chem. Soc. 1993, 8116. Martin J. Am. Chem. Soc. 2001, 8003. Overman J. Am. Chem. Soc. 1995, 5776. Bodwell Angew. Chem. Int. Ed. 2002, 3261. Kuehne J. Org. Chem. 1993, 7490. Mori J. Am. Chem. Soc. 2003, 9801. Kuehne J. Org. Chem. 1998, 9427. Shibasaki Tetrahedron 2004, 9569. Rawal J. Org. Chem. 1994, 2685. Fukuyama J. Am. Chem. Soc. 2004, 10246. Bosch, Bonjoch Chem. Eur. J. 2000, 655. Padwa Org. Lett. 2007, 279. History and Structure of (!)-Strychnine ! Isolated in pure form in 1818 (Pelletier and Caventou) ! Structural Determination in 1947 (Robinson and Leuchs) ! 24 skeletal atoms (C21H22N2O2) ! Isolated in pure form in 1818 (Pelletier and Caventou) N ! Over !25 07 -pruinbglisc,a 6ti-osntesr epoecrteaninteinrgs to structure ! Structural Determination in 1947 (Robinson and Leuchs) ! Notor!io u Ss ptoirxoicne (nletethr a(lC d-o7s) e ~10-50 mg / adult) N H H ! Over 250 publications pertaining to structure O O ! Comm!o nClyD uEs eridn gr osdyesntet mpoison ! Notorious poison (lethal dose ~10-50 mg / adult) ! Hydroxyethylidine Strychnos nux vomica ! $20.20 / 10 g (Aldrich), ~1.5 wt% (seeds), ~1% (blossoms) "For it's molecular size it is the most complex substance known." -Robert Robinson History and Structure of (!)-Strychnine ! 24 skeletal atoms (C21H22N2O2) N ! 7-rings, 6-stereocenters ! Spirocenter (C-7) N H H O O ! CDE ring system ! Hydroxyethylidine "For it's molecular size it is the most complex substance known." -Robert Robinson "If we can't make strychnine, we'll take strychnine" -R. -

L. Lewis Acid Catalysis
L. LEWIS ACID CATALYSIS Background Twenty amino acids is not enough. The breadth of chemistry handled by enzymes requires that additional chemical species be employed in catalysis. So-called cofactors are non-amino acid components of enzymes that may be either associated or bonded to proteins and contribute to rate acceleration. Roughly one-third of all enzymes studied to date employ metal ion cofactors. Metal cations can play one of two roles; they may act as Lewis acids or as redox reagents. As Lewis acids, they function in a role similar to general acids, stabilizing negative charge in the enzyme active site. This chapter will focus on Lewis acid catalysis in reactions that employ water as a nucleophile. Lewis acid catalysis can be observed to play a role in either (a) activating water as a nucleophile or (b) activating the electrophile for nucleophilic attack. Activation of Water as a Nucleophile As noted earlier, water is a poor nucleophile due to the build up of positive charge on the oxygen that accompanies attack upon an electrophile. General base catalysis overcomes that issue by promoting deprotonation of the water molecule as it attacks. Another approach is to stabilize the hydroxide ion within the enzyme active site. While the metal ion itself is a Lewis acid (electron pair acceptor), the hydrated metal ion is a Bronsted acid (a proton donor) according to Equation L.1. n+ (n-1)+ + M(H2O)6 ⇔ M(H2O)5(OH) + H Eq L.1 In this instance Lewis acidity and Bronsted acidity are linked. The capacity of the metal cation to stabilize negative charge from a hydroxide (hydroxo in inorganic-speak) ligand is a reflection of the Lewis acidity of the cation. -
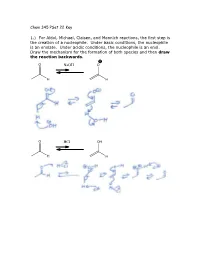
Chem 345 Pset 22 Key 1.) for Aldol, Michael, Claisen, and Mannich
Chem 345 PSet 22 Key 1.) For Aldol, Michael, Claisen, and Mannich reactions, the first step is the creation of a nucleophile. Under basic conditions, the nucleophile is an enolate. Under acidic conditions, the nucleophile is an enol. Draw the mechanism for the formation of both species and then draw the reaction backwards. O NaOH O H H O HCl OH H H 2.) In the case of a Mannich reaction, the electrophile (an imine) must also be made. Draw the mechanism for the forward and backward reaction: O MeNH2 N H H cat. AcOH H H 3.) When a secondary amine is used and the carbonyl has an enolizable proton, an enamine can be formed. Are enamines nucleophilic or electrophilic? Nucleophilic H O N N H2O H cat. AcOH H enamine N N H H enamine 4.) The following reaction is the Lobry-de-Bruyn-von-Ekenstein reaction. Don’t worry about the name. I only recently found out that that was the official name for the rearrangement leading from glucose to fructose or mannose and back again. I was taught it under a different name: the Carbohydrate Game. Under acidic or basic conditions, it is possible that the following sugars (glucose, fructose, and mannose) are in equilibrium with each other. Draw the acid catalyzed version and the base catalyzed version. (Hint: this is just a repeat of question 1.) O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose The sugars are drawn in a Fisher projection (or what I think of is a dead cat projection).