Addition/Nitro-Mannich Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Abstract Multicomponent Reactions of Salicylaldehyde, Cyclic Ketones, and Arylamines Through Cooperative Enamine-Metal Lewis
ABSTRACT MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS by Ryan Gregory Sarkisian Multicomponent reactions (MCRs) are the most atom economic, highly selective, and convergent type of reaction. This allows for a reaction to have a wide scope and allows for maximization of the complexity of a product. Catalyzing these MCRs with asymmetric catalysis is a novel way to introduce stereocontrol into highly complex molecules with various functional groups. Asymmetric catalysis is considered the most efficient method for constructing highly functionalized optically active stereopure compounds. There are three pillars of asymmetric catalysis: biocatalysis, transition metal catalysis, and organocatalysis. This research focuses on two of these pillars, transition metal catalysis and organocatalysis, working cooperatively to catalyze this MCR. The focus is to educate or refresh the audience on the basic topics that make up the complexity of the MCRs being catalyzed by cooperative asymmetric catalysis. Ultimately to explore the cooperative catalysts used to synthesize both the racemic and asymmetric three fused ring products (9-((4-methoxyphenyl)amino)-2,3,4,4a,9,9a-hexahydro-1H-xanthen-4a-ol). MULTICOMPONENT REACTIONS OF SALICYLALDEHYDE, CYCLIC KETONES, AND ARYLAMINES THROUGH COOPERATIVE ENAMINE-METAL LEWIS ACID CATALYSIS A THESIS Submitted to the Faculty of Miami University in partial fulfillment of the requirements for the degree of Master of Science Department of -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction
Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction Mary A. Yohannan* and Arthur Berrier U.S. Department of Justice Drug Enforcement Administration Special Testing and Research Laboratory 22624 Dulles Summit Court Dulles, VA 20166 [email: mary.a.yohannan -at- usdoj.gov] ABSTRACT: The formation of imine by-products from phenethylamines, amphetamines, and tryptamines upon an acetone extraction is presented. These imine by-products were characterized using GC/MSD and exhibited preferential cleavage at the α-carbon of the alkyl chain. Further characterization of the imine by-products of phenethylamine and tryptamine was done using IR and NMR. KEYWORDS: phenethylamine, tryptamine, imine, acetone, schiff base, drug chemistry, forensic chemistry In most forensic laboratories, the solvents used to extract at the α-carbon on the alkyl chain. In addition to GC/MS, the drugs are chosen based upon their solubility properties and their imines formed from phenethylamine base and tryptamine base ability to not interact with the drug. In fact, there are very few were characterized by Fourier transform-infrared spectroscopy publications where a solvent used to extract a drug reacts with (FTIR) and nuclear magnetic resonance (NMR) spectroscopy. the drug and forms by-products [1-3]. This laboratory recently discovered that an additional Experimental component was formed when acetone was used to extract a Solvents, Chemicals, and Materials sample containing a known tryptamine. Analysis by gas Acetone was ACS/HPLC grade from Burdick and Jackson chromatography/mass spectroscopy (GC/MS) of the acetone Laboratories (Muskegon, MI). Phenethylamine base and extract yielded an extra peak in the total ion chromatogram that tryptamine base were obtained from Sigma-Aldrich Chemicals was approximately half the abundance of the known tryptamine (Milwaukee, WI). -

Comparative Total Syntheses of Strychnine
Comparative Total Syntheses of Strychnine N C D MacMillan Group Meeting N H O H A H B E Nathan Jui H N N H H F G July 22, 2009 O O O References: Pre-Volhardt: Bonjoch Chem. Rev. 2000, 3455. Woodward Tetrahedron, 1963, 247. Volhardt J. Am. Chem. Soc. 2001, 9324. Magnus J. Am. Chem. Soc. 1993, 8116. Martin J. Am. Chem. Soc. 2001, 8003. Overman J. Am. Chem. Soc. 1995, 5776. Bodwell Angew. Chem. Int. Ed. 2002, 3261. Kuehne J. Org. Chem. 1993, 7490. Mori J. Am. Chem. Soc. 2003, 9801. Kuehne J. Org. Chem. 1998, 9427. Shibasaki Tetrahedron 2004, 9569. Rawal J. Org. Chem. 1994, 2685. Fukuyama J. Am. Chem. Soc. 2004, 10246. Bosch, Bonjoch Chem. Eur. J. 2000, 655. Padwa Org. Lett. 2007, 279. History and Structure of (!)-Strychnine ! Isolated in pure form in 1818 (Pelletier and Caventou) ! Structural Determination in 1947 (Robinson and Leuchs) ! 24 skeletal atoms (C21H22N2O2) ! Isolated in pure form in 1818 (Pelletier and Caventou) N ! Over !25 07 -pruinbglisc,a 6ti-osntesr epoecrteaninteinrgs to structure ! Structural Determination in 1947 (Robinson and Leuchs) ! Notor!io u Ss ptoirxoicne (nletethr a(lC d-o7s) e ~10-50 mg / adult) N H H ! Over 250 publications pertaining to structure O O ! Comm!o nClyD uEs eridn gr osdyesntet mpoison ! Notorious poison (lethal dose ~10-50 mg / adult) ! Hydroxyethylidine Strychnos nux vomica ! $20.20 / 10 g (Aldrich), ~1.5 wt% (seeds), ~1% (blossoms) "For it's molecular size it is the most complex substance known." -Robert Robinson History and Structure of (!)-Strychnine ! 24 skeletal atoms (C21H22N2O2) N ! 7-rings, 6-stereocenters ! Spirocenter (C-7) N H H O O ! CDE ring system ! Hydroxyethylidine "For it's molecular size it is the most complex substance known." -Robert Robinson "If we can't make strychnine, we'll take strychnine" -R. -
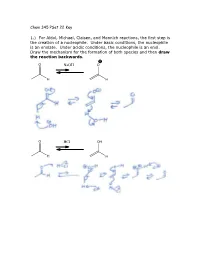
Chem 345 Pset 22 Key 1.) for Aldol, Michael, Claisen, and Mannich
Chem 345 PSet 22 Key 1.) For Aldol, Michael, Claisen, and Mannich reactions, the first step is the creation of a nucleophile. Under basic conditions, the nucleophile is an enolate. Under acidic conditions, the nucleophile is an enol. Draw the mechanism for the formation of both species and then draw the reaction backwards. O NaOH O H H O HCl OH H H 2.) In the case of a Mannich reaction, the electrophile (an imine) must also be made. Draw the mechanism for the forward and backward reaction: O MeNH2 N H H cat. AcOH H H 3.) When a secondary amine is used and the carbonyl has an enolizable proton, an enamine can be formed. Are enamines nucleophilic or electrophilic? Nucleophilic H O N N H2O H cat. AcOH H enamine N N H H enamine 4.) The following reaction is the Lobry-de-Bruyn-von-Ekenstein reaction. Don’t worry about the name. I only recently found out that that was the official name for the rearrangement leading from glucose to fructose or mannose and back again. I was taught it under a different name: the Carbohydrate Game. Under acidic or basic conditions, it is possible that the following sugars (glucose, fructose, and mannose) are in equilibrium with each other. Draw the acid catalyzed version and the base catalyzed version. (Hint: this is just a repeat of question 1.) O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose O O H H H2C OH + + HO H H3O H OH H3O O or or HO- HO- HO H HO H HO H H OH H OH H OH H OH H OH H OH H2C OH H2C OH H2C OH D-glucose D-fructose D-mannose The sugars are drawn in a Fisher projection (or what I think of is a dead cat projection). -

The Carcinogenicity of the O-Methoxy Derivatives of N-2-Fluorenylacetamide and of Related Compounds in the Rat
[CANCER RESEARCH 28, 234-244, February 1968] The Carcinogenicity of the o-Methoxy Derivatives of N-2-Fluorenylacetamide and of Related Compounds in the Rat H. R. Gutmann, S. B. Galitski, and W. A. Foley Laboratory ]or Cancer Research, Veterans Administration Hospital, and Department o] Biochemistry, University o] Minnesota, Minneapolis, Minnesota 55417 SUMMARY acetamide by the sequential reactions of deacetylation and oxidation. In order to test the idea that the lack of carcinogenicity of the o-amidofluorenols, N- (1-hydroxy-2-fluorenyl) acetamide and INTRODUCTION N-(3-hydroxy-2-fluorenyl)acetamide, is due to the hydrophilic phenolic hydroxyl group, the methylated derivatives, N-(1- Several model studies from this laboratory have shown that methoxy-2-fluorenyl)acetamide and N-(3-methoxy-2-fluorenyl) the o-quinone imines, 2-imino-l,2-fluorenoquinone and 2-imino- acetamide as well as the hydrochlorides of 1-methoxy-2-fluo- 2,3-fluorenoquinone, which are derived from the carcinogen renamine and 3-methoxy-2-fiuorenamine, were prepared, and N-2-fluorenylacetamide by the sequential enzymatic reactions their carcinogenicity was evaluated in the rat. N-(1-Methoxy- of hydroxylation, deacetylation, and oxidation (6, 10, 12, 27, 2-fluorenyl)acetamide and 1-methoxy-2-fluorenamine hydro- 34, 35), form stable adducts with a variety of proteins (17, 18). chloride, when administered orally to male rats for 5 months, However, the relevance of the binding of these o-quinone imines gave a tumor incidence of 27 and 50%, respectively. Approxi- to chemical carcinogenesis has remained obscure largely be- mately one-half of the lesions produced by either com- cause the o-amidofluorenols, 1-OH-AAF 2 and 3-OH-AAF pound were adenocarcinomas of the small intestine. -

Synthesis and Characterization of Novel Imine-Linked Covalent Organic Frameworks
Synthesis and Characterization of Novel Imine-Linked Covalent Organic Frameworks THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Toni Beirl Graduate Program in Chemistry The Ohio State University 2015 Master's Examination Master's Examination Committee: Professor Psaras McGrier, Advisor Professor Jovica D. Badjic Copyrighted by Toni M.Beirl 2015 Abstract Covalent organic frameworks (COFs) are a class of porous crystalline materials composed of light elements (such as H, B, C, N, and O) that are linked by covalent bonds. The modular nature of COFs permits the integration of various π-conjugated molecular building blocks into highly ordered polymeric structures with low densities and high thermal stabilities making them suitable for applications related to energy storage and conversion, catalysis, and gas storage. Since a majority of the early examples of COFs contained boroxine or boronate esters linkages, many of these materials were often susceptible to hydrolysis when exposed to aqueous conditions resulting in decomposition of the framework. The recent discovery of imine-linked COFs has sparked the creation of COFs with superior chemical stability on account of an intramolecular hydrogen bond between the hydroxyl and imine functional groups, which enhances their stability in aqueous and acidic environments. Utilizing this feature, this thesis examines the synthesis and gas adsorption properties of novel imine-linked COFs that contain 1,3,5-tris(styryl)benzene and 1,3,5– tris(arylethynyl)benzene π-conjugated units. By creating analogs which were fluorescent in both solution and solid-state, studies were conducted to determine their ability to serve as chemical sensors for explosives. -

Modeling Nitrile-Terminated Polypropylene Imine Dendrimer Fragmentation with DFT
Modeling Nitrile-Terminated Polypropylene Imine Dendrimer Fragmentation with DFT Eric W. Martin, Jacob T. Kilgore and William D. Price Marshall University Department of Chemistry Huntington, WV 25755 Abstract Dendrimers are regularly branched polymers with a treelike structure that can be tuned for size, shape, and functionality. This relatively new class of compound has shown potential for useful host-guest chemistries including site-specific drug delivery via molecular recognition, catalysis, and nonlinear optics. Gas-phase dissociation studies have been initiated to probe the structure and stability of the half and first generation polypropylene dendrimer complexes and to develop an analytical framework for their characterization. These dissociation studies result in fragmentation products of mass-to-charge ratios that can be assigned to multiple possible isomers formed by potentially competing mechanisms. Since these reactions are under kinetic control we will present density functional results for modeling the dissociation mechanisms for the most abundantly produced fragments from the protonated dendrimers. The BMK functional in conjunction with a moderately-sized basis has been chosen for its utility in determining transition state energies and kinetic parameters. Introduction Dendrimers have several promising and exciting possibilities ranging from use as chelating agents1 to site-specific host-guest chemistry and controlled gene2 and drug delivery systems3. Distinct properties of dendrimers, such as well-defined architecture and a high ratio of moieties to molecular volume, make these materials highly interesting for the development of nanomaterials and medicines4. The compact, nanometer- scale structure of the hyperbranched polymer results in high solubility and low solution viscosity5, while the dense arrangement of functional groups allows pharmaceutical agents and magnetic resonance imaging contrast dyes to be chemically grafted to the dendrimer in concentrated amounts6. -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics