Enolate Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Optimizing the Least Nucleophilic Anion. a New, Strong Methyl+ Reagent Daniel Stasko and Christopher A
Published on Web 01/25/2002 Optimizing the Least Nucleophilic Anion. A New, Strong Methyl+ Reagent Daniel Stasko and Christopher A. Reed* Department of Chemistry, UniVersity of California, RiVerside, California 92521-0304 Received August 3, 2001 The ideal weakly coordinating counterion for reactive cations would be the least nucleophilic, least basic, most inert anion available. It should also be inexpensive, have useful spectroscopic handles, and confer good solubility and crystallizing properties on - its salts. Triflate anion, CF3SO3 , has served chemistry well in this regard but larger size, absence of lone pairs, and extreme chemical inertness have recently made carboranes1 or fluorinated tetraphe- nylborates the preferred choice for many applications.2 These anions have led to the solution of the silylium ion problem,3 enhanced Lewis acid catalysis by Li+ ion,4-6 new “strong-but-gentle” superacids,7 commercially viable olefin polymerization catalysts,8 new possibilities for electrolytes9,10 and ionic liquids,11 and the •+ 7 + 12 isolation of new reactive cations such as C60 , Bu3Sn , Cu- + 13 (CO)4 , etc. Figure 1. The 1-H-2,3,4,5,6-pentamethyl-7,8,9,10,11,12-hexahalo-CB11 - ) The perfluorinated tetraphenylborate anion is the preferred choice carborane anions, 1-H-CB11Me5X6 (X Cl, Br, I), used in this work. for price and solubility reasons but its salts frequently form liquid 29 Table 1. Selected Si Chemical Shifts (ppm) for i-Pr3SiY clathrates and fail to crystallize. The boron-phenyl bond is rather 29 easily cleaved by strong Brønsted14 or Lewis15 acids. Halogenated compd Si conditions carborane anions are much more inert than tetraphenylborates, their i-Pr3Si(1-H-CB11H5Br6) 100 C6D6 salts crystallize well, but they are more expensive and their salts i-Pr3Si(1-H-CB11H5Cl6) 103 C6D6 i-Pr Si(F -BPh )a 108 solid state less soluble. -
Ochem ACS Review 18 Enols and Enolates
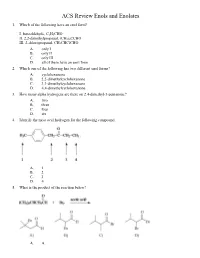
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Tautomer Identification and Tautomer Structure Generation Based on The
J. Chem. Inf. Model. 2010, 50, 1223–1232 1223 Tautomer Identification and Tautomer Structure Generation Based on the InChI Code Torsten Thalheim,†,‡ Armin Vollmer,† Ralf-Uwe Ebert,† Ralph Ku¨hne,† and Gerrit Schu¨u¨rmann*,†,‡ UFZ Department of Ecological Chemistry, Helmholtz Centre for Environmental Research, Permoserstrasse 15, 04318 Leipzig, Germany, and Institute for Organic Chemistry, Technical University Bergakademie Freiberg, Leipziger Strasse 29, 09596 Freiberg, Germany Received March 27, 2010 An algorithm is introduced that enables a fast generation of all possible prototropic tautomers resulting from the mobile H atoms and associated heteroatoms as defined in the InChI code. The InChI-derived set of possible tautomers comprises (1,3)-shifts for open-chain molecules and (1,n)-shifts (with n being an odd number >3) for ring systems. In addition, our algorithm includes also, as extension to the InChI scope, those larger (1,n)-shifts that can be constructed from joining separate but conjugated InChI sequences of tautomer-active heteroatoms. The developed algorithm is described in detail, with all major steps illustrated through explicit examples. Application to ∼72 500 organic compounds taken from EINECS (European Inventory of Existing Commercial Chemical Substances) shows that around 11% of the substances occur in different heteroatom-prototropic tautomeric forms. Additional QSAR (quantitative structure-activity relationship) predictions of their soil sorption coefficient and water solubility reveal variations across tautomers up to more than two and 4 orders of magnitude, respectively. For a small subset of nine compounds, analysis of quantum chemically predicted tautomer energies supports the view that among all tautomers of a given compound, those restricted to H atom exchanges between heteroatoms usually include the thermodynamically most stable structures. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Lecture 6 the Crossed Aldol Reaction and Its Many Variants
Lecture 6 The Crossed Aldol Reaction and its Many Variants Objectives: By the end of this lecture you will be able to: 1. make an appropriate choice of base to completely enolise carbonyl compounds; 2. use enolates in a crossed aldol reaction; 3. recognise the aldol functional group motif, and its variants, in complex molecules; 4. use the aldol disconnection to simplify a retrosynthetic analysis; 5. use the Reformatski and Mukaiyama aldol reactions in synthesis; 6. draw arrow-pushing mechanisms for all the aldol reactions discussed in this lecture. Introduction We have seen how choosing a base (B-) whose conjugate acid (B−H) is a much poorer acid − i.e. has a much higher pKa − than the proton we wish to abstract, ensures effectively complete deprotonation of the α-C−H of a carbonyl compound to provide the corresponding enolate. By completely enolising the carbonyl group, we can suppress self-condensation aldol processes, and instead use a different electrophile such as an aldehyde to form a β-hydroxy carbonyl compound. This reaction sequence is identical in basic mechanism to the intramolecular aldol processes that we discussed in previous lectures. It is now described as a crossed aldol condensation because the electrophile is a different carbonyl compound to the one that was used to form the nucleophilic enolate. This reaction, and its many variants, provides one of the most important methods for preparing C−C bonds. Pattern Recognition The aldol reaction and its many variants are very useful reactions in synthesis. You need to be able to identify the patterns or functional group motifs where this type of bond-forming process can be used. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -
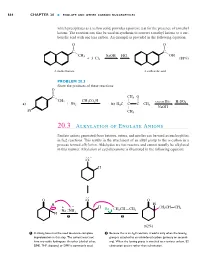
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Determination of Solvent Effects on Ketoðenol Equilibria of 1,3-Dicarbonyl Compounds Using NMR: Revisiting a Classic Physical C
In the Laboratory Determination of Solvent Effects on Keto–Enol Equilibria W of 1,3-Dicarbonyl Compounds Using NMR Revisiting a Classic Physical Chemistry Experiment Gilbert Cook* and Paul M. Feltman Department of Chemistry, Valparaiso University, Valparaiso, IN 46383; *[email protected] “The influence of solvents on chemical equilibria was discovered in 1896, simultaneously with the discovery of keto–enol tautomerism in 1,3-dicarbonyl compounds” (1). The solvents were divided into two groups according to their ability to isomerize compounds. The study of the keto–enol tautomerism of β-diketones and β-ketoesters in a variety of solvents using proton NMR has been utilized as a physical Figure 1. The β-dicarbonyl compounds studied in the experiment. chemistry experiment for many years (2, 3). The first reported use of NMR keto–enol equilibria determination was by Reeves (4). This technique has been described in detail in an experiment by Garland, Nibler, and Shoemaker (2). panded (i) to give an in-depth analysis of factors influencing The most commonly used β-diketone for these experi- solvent effects in tautomeric equilibria and (ii) to illustrate ments is acetylacetone (Scheme I). Use of proton NMR is a the use of molecular modeling in determining the origin of viable method for measuring this equilibrium because the a molecule’s polarity. The experiment’s original benefits of tautomeric keto–enol equilibrium is slow on the NMR time using proton NMR as a noninvasive method of evaluating scale, but enol (2a)–enol (2b) tautomerism is fast on this scale equilibrium are maintained. (5). It has been observed that acyclic β-diketones and β- Experimental Procedure ketoesters follow Meyer’s rule of a shift in the tautomeric equi- librium toward the keto tautomer with increasing solvent Observations of the solvent effects for three other 1,3- polarity (6). -

Mechanisms of Carbonyl Activation by BINOL N-Triflylphosphoramides
Mechanisms of Carbonyl Activation by BINOL N-Triflylphosphoramides Enantioselective Nazarov Cyclizations Lovie-toon, Joseph P.; Tram, Camilla Mia; Flynn, Bernard L.; Krenske, Elizabeth H. Published in: ACS Catalysis DOI: 10.1021/acscatal.7b00292 Publication date: 2017 Document version Publisher's PDF, also known as Version of record Citation for published version (APA): Lovie-toon, J. P., Tram, C. M., Flynn, B. L., & Krenske, E. H. (2017). Mechanisms of Carbonyl Activation by BINOL N-Triflylphosphoramides: Enantioselective Nazarov Cyclizations . ACS Catalysis, 7(5), 3466-3476. https://doi.org/10.1021/acscatal.7b00292 Download date: 09. Apr. 2020 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Research Article pubs.acs.org/acscatalysis Mechanisms of Carbonyl Activation by BINOL N‑Triflylphosphoramides: Enantioselective Nazarov Cyclizations Joseph P. Lovie-Toon,† Camilla Mia Tram,†,‡,∥ Bernard L. Flynn,§ and Elizabeth H. Krenske*,† † School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia ‡ University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark § Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Victoria 3052, Australia *S Supporting Information ABSTRACT: BINOL N-triflylphosphoramides are versatile organocatalysts for reactions of carbonyl compounds. Upon activation -

Probing Ion Exchange in the Triflic Acid-Guanidinium Triflate System: a Solid- State Nuclear Magnetic Resonance Study
Probing ion exchange in the triflic acid-guanidinium triflate system: a solid- state nuclear magnetic resonance study Citation: Zhu, Haijin, MacFarlane, Douglas and Forsyth, Maria 2014, Probing ion exchange in the triflic acid-guanidinium triflate system: a solid-state nuclear magnetic resonance study, Journal of Physical Chemistry C, vol. 118, no. 49, pp. 28520-28526. This is the accepted manuscript. ©2014, American Chemical Society This document is the Accepted Manuscript version of a Published Work that appeared in final form in Journal of Physical Chemistry C, copyright © American Chemical Society after peer review and technical editing by the publisher. To access the final edited and published work see http://pubs.acs.org/doi/abs/10.1021/jp5101472 Downloaded from DRO: http://hdl.handle.net/10536/DRO/DU:30070195 DRO Deakin Research Online, Deakin University’s Research Repository Deakin University CRICOS Provider Code: 00113B Page 1 of 19 The Journal of Physical Chemistry 1 2 3 4 Probing Ion Exchange in the Triflic Acid - Guanidinium Triflate System: a 5 6 Solid-State NMR Study 7 8 9 Haijin Zhu,*,a Douglas MacFarlaneb and Maria Forsytha 10 11 a Institute for Frontier Materials and the ARC Centre of Excellence for Electromaterials 12 Science, Deakin University, Geelong, VIC 3216, Australia. E-mail: [email protected]; 13 Fax: 61 (03) 92446868; Tel: +61 (03) 52273696 14 15 b Department of Chemistry and the ARC Centre of Excellence for Electromaterials 16 Science, Monash University, Clayton, VIC 3800, Australia 17 18 19 * Corresponding author, Tel: 61 (03) 52273696; Fax: 61 (03) 92446868; Email: 20 [email protected] (H.