20.3 Alkylation of Enolate Anions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alkyl Halides Work Best; Secondary Alkyl Halides Give Lower Yields
CHAPTER 21 ESTER ENOLATES ou have already had considerable experience with carbanionic compounds and their applications in synthetic organic chemistry. The first was acetylide ion in YChapter 9, followed in Chapter 14 by organometallic compounds—Grignard reagents, for example—that act as sources of negatively polarized carbon. In Chapter 18 you learned that enolate ions—reactive intermediates generated from aldehydes and ketones—are nucleophilic, and that this property can be used to advantage as a method for carbon–carbon bond formation. The present chapter extends our study of carbanions to the enolate ions derived from esters. Ester enolates are important reagents in synthetic organic chemistry. The stabilized enolates derived from -keto esters are particularly useful. OO C ␣ C  R CH2 ORЈ -Keto ester: a ketone carbonyl is  to the carbonyl group of the ester. A proton attached to the ␣-carbon atom of a -keto ester is relatively acidic. Typical Ϫ11 acid dissociation constants Ka for -keto esters are Ϸ10 (pKa 11). Because the ␣- carbon atom is flanked by two electron-withdrawing carbonyl groups, a carbanion formed at this site is highly stabilized. The electron delocalization in the anion of a -keto ester is represented by the resonance structures 831 832 CHAPTER TWENTY-ONE Ester Enolates Ϫ Ϫ O O O O O O C C C C C C R C ORЈ R ϪC ORЈ R C ORЈ H H H Principal resonance structures of the anion of a -keto ester We’ll begin by describing the preparation and properties of -keto esters, proceed to a discussion of their synthetic applications, continue to an examination of related species, and conclude by exploring some recent developments in the active field of synthetic car- banion chemistry. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Carboxylic Acids and Their Derivatives
8 CARBOXY A lmost all of the basic types of reactions now have been covered: addition, elimination, substitution, and rearrangement by polar, radical, and concerted mechanisms. Indeed, if you have been looking for similarities, you will have seen that most of the reactions discussed in the preceding three chapters are variations on basic types we have discussed earlier. Furthermore, most of the basic structural effects that determine chemical reactivity also have been covered in previous chapters: bond energies, steric hindrance, electronega- tivity, electron delocalization, hydrogen bonding, solvation, and conforma- tional influences. You might well ask what is left. The answer is, a great deal- but now we will be concerned mostly with putting concepts together, moving from the simple to the complex. For example, in this chapter we will be trying to under- stand the ways that carboxylic acids, which possess the -c' functional \ group, are similar to and different from alcohols, which have the OH group, and aldehydes and ketones, which have C=O bonds. Subsequently we will look at acids that also possess OH or NH, sub- stituent groups (or both) and develop a rationale for the behavior of these combinations in terms of effects we already have discussed. Insofar as pos- sible, you should try to do this yourself whenever you encounter a substance with a new set of combinations of functional groups on its molecules. You often will be in error (as many experts will be), because even if you take Carboxylic Acids and Their Derivatives 789 account of all of the structural effects, as well as the possible reactions or inter- actions, the overall result of these frequently is very difficult to judge in advance. -

Carbonyl Condensation Reactions
24 Carbonyl Condensation Reactions 24.1 The aldol reaction 24.2 Crossed aldol reactions 24.3 Directed aldol reactions 24.4 Intramolecular aldol reactions 24.5 The Claisen reaction 24.6 The crossed Claisen and related reactions 24.7 The Dieckmann reaction 24.8 The Michael reaction 24.9 The Robinson annulation Ibuprofen is the generic name for the pain reliever known by the trade names of Motrin and Advil. Like aspirin, ibuprofen acts as an anti-infl ammatory agent by blocking the synthesis of prostaglandins from arachidonic acid. One step in a commercial synthesis of ibuprofen involves the reaction of a nucleophilic enolate with an electrophilic carbonyl group. In Chap- ter 24, we learn about the carbon–carbon bond-forming reactions of enolates with carbonyl electrophiles. 916 smi75625_ch24_916-948.indd 916 11/12/09 12:12:52 PM 24.1 The Aldol Reaction 917 In Chapter 24, we examine carbonyl condensations—that is, reactions between two car- bonyl compounds—a second type of reaction that occurs at the α carbon of a carbonyl group. Much of what is presented in Chapter 24 applies principles you have already learned. Many of the reactions may look more complicated than those in previous chapters, but they are fundamen- tally the same. Nucleophiles attack electrophilic carbonyl groups to form the products of nucleo- philic addition or substitution, depending on the structure of the carbonyl starting material. Every reaction in Chapter 24 forms a new carbon–carbon bond at the ` carbon to a carbonyl group, so these reactions are extremely useful in the synthesis of complex natural products. -
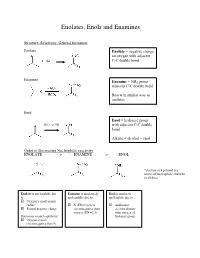
Enolates, Enols and Enamines
Enolates, Enols and Enamines Structure, Reactivity, General formation Enolate Enolate = negative charge on oxygen with adjacent C-C double bond Enamine Enamine = NR2 group adjacent C-C double bond Reacts in similar way as enolates Enol Enol = hydroxyl group - H3O+ or OH with adjacent C-C double bond Alkene + alcohol = enol Order of Decreasing Nucleophilic reactivity ENOLATE > ENAMINE > ENOL *electron-rich pi bond is a source of nucleophilic character in all three Enolate is nucleophilic due Enamine is moderately Enol is moderate to… nucleophilic due to… nucleophile due to… Oxygen’s small atomic radius N (EN=3) is less Additional Formal negative charge electronegative than electron density oxygen (EN =3.5) from oxygen of Detractors to nucleophilicity: hydroxyl group Oxygen is more electronegative than N When looking at which side is favored (products or reactants): Equilibrium favors side with WEAKER acid/base pair Weakest acid and weakest base should be on SAME side When comparing pKa values, juxtapose just the acids or just the bases Side with larger pka value (weaker acid) favored in equilibrium Remember that differences in pKa values reflect a difference quantity of 10x Example: compound H-A (pKa=3) + base compound A (pKa=10) + H-base 7 equilibrium lies to the RIGHT by factor of 10 Some Useful pKa values (from more acidic basic) H2SO4 (pka = -9) H2O (pka = 15.7) + H3O (pka = -1.8) CH3COCH3 (pka = 19) B-diketone (pka = 9) CH3COOCH3 (pKa = 25) HCN (pka = 9.1) LDA-H (pka = 36) CH3OH (pka = 15.5) How do we know which proton to deprotonate? Deprotonate most acidic proton usually leads to the most stable enolate 1. -

Chap 11. Carbonyl Alpha-Substitution Reactions and Condensation Reactions
Chap 11. Carbonyl Alpha-Substitution Reactions and Condensation Reactions Four fundamental reactions of carbonyl compounds 1) Nucleophilic addition (aldehydes and ketones) 2) Nucleophilic acyl substitution (carboxylic acid derivatives) 3) The alpha-substitution Reactions occur at the position next to the carbonyl group–the alpha position–and result in the substitution of an α hydrogen atom by some other electrophilic group, E+: O O E+ α H E + H+ 4) The carbonyl condensation Reactions take place when two carbonyl compounds react with each other in such a way that the α carbon of one partner becomes bonded to the carbonyl carbon of the second partner. O H O OH 2 H The key feature of the reactions (3, 4) is that they take place through the formation of either enol or enolate ion intermediates. H O O C C C C 11.1 Keto-Enol Tautomerism The interconversion between the keto and enol forms of carbonyl compounds is a special kind of isomerism called tautomerism. In Greek, tauto means “the same,” and meros means “part.” The interconversion of tautomers involves the movement of atoms (H), so they are different from resonance forms. H O Rapid O α H Keto tautomer Enol tautomer 1 Most carbonyl compounds exist almost entirely in the keto form at equilibrium. OOHO OH 99.9999% 0.0001% 99.999999% 0.000001% Mechanism of keto-enol tautomerism Acid catalysis H A H H H O O O A O H H H + HA Keto tautomer Enol tautomer Base catalysis HOH H OOOH OO H + OH Keto tautomer Enol tautomer 11.2 Reactivity of Enols: The Mechanism of Alpha-Substitution Reactions Since their double bonds are electron-rich, enols behave as nucleophiles and react with electrophiles in the same way as alkenes do. -

Enolate Chemistry
Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 1 Enolate Chemistry 1. Some Basics In most cases the equilibrium lies almost completely on the side of the ketone. The ketone tautomer is electrophilic and reacts with nucleophiles: The enol tautomer is nucleophilic and reacts with electrophiles. There are two possible products - enols are ambident nucleophiles: The nucleophilic enol tautomer (and especially the enolate variant) is one of the most important reactive species for C-C bond formation. Treat a ketone with an appropriate base and can get deprotonation at the α-position to form an enolate: Enolates are synthetically much more useful than enols (although they react analogously). Imine anions and eneamines are synthetic equivalents of enolate anions. Li N N H H Imine anion eneamine By knowing the pKa values of the relevant acidic protons it is possible to predict suitable bases for forming the corresponding enolates. Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 2 Some Important pKa Values - - - O O HC N HC3 NO2 H2CN+ 2 + O O pKa ~ 10 - Enolates are nucleophiles and ketones are electrophiles - therefore there is always the potential problem for self condensation. If this is desirable we need to use a base which does not completely deprotonate the carbonyl compound i.e. set up an equilibrium. This is best achieved when the pKa of the carbonyl group and conjugate acid (of the base) are similar: Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 3 pKa (tBuOH) ~ 18. A stronger base. pKa difference of 2. Ratio of ketone to enolate will be of the order 100:1 i.e. -

The Α-Carbon Atom and Its Pka the Inductive Effect of the Carbonyl
Chapter 18: Enols and Enolates O E+ O H Enolate O E carbonyl H O E+ Enol 18.1: The α-Carbon Atom and its pKa O "' " #' !' ! # 126 The inductive effect of the carbonyl causes the α-protons to be more acidic. The negative charge of the enolate ion (the conjugate base of the aldehyde or ketone) is stabilized by resonance delocalization. The pKa of the α-protons of aldehydes and ketones is in the range of 16-20 (Table 18.1, p. 754) resonance effect ! - inductive O O !+ O + B effect C H C C + H-B !+ C C C carbonyl Enolate anion H C C C C C C O O O O H3C CH3 C H3CH2COH H3C CH3 ethane acetone ethanol 127 pKa= 50-60 pKa= 19 pKa= 16 65 O O O O O C C Cl C C C H3C CH3 H3C C H3C C H3C C CH3 H H H H H H pKa 19 14 16 9 O O + H O C + H3O C 2 H C CH H3C CH3 3 2 acid base conjugate conjugate base acid pKa 19 -1.7 (weaker acid) (weaker base) (stronger base) (stronger acid) O O + HO C + H O C H C CH 2 H3C CH3 3 2 acid base conjugate conjugate base acid pKa 19 15.7 (weaker acid) (weaker base) (stronger base) (stronger acid) 128 O O O O C C + H2O C C + HO H C C CH H C C CH 3 3 3 3 H H H acid base conjugate conjugate base acid pKa 9 15.7 (stronger acid) (stronger base) (weaker base) (weaker acid) 18.2: The Aldol Condensation- An enolate of one carbonyl (nucleophile) reacts with the carbonyl carbon (electrophile) of a second carbonyl compound resulting in the formation of a new C-C bond O OH O 2 H C C H + HO + HO 3 H3C CH CH2 C H acetaldehyde 3-hydroxybutanal (!-hydroxy aldehyde) 129 66 Mechanism of the base-catalyzed aldol condensation (Fig. -

Chem 353: Aldol Condensation
ALDOL.1 ORGANIC SYNTHESIS: ALDOL CONDENSATION REACTION TECHNIQUES REQUIRED: Filtration (Vacuum), Recrystallisation, Melting Point Determination, Yield calculation OTHER DOCUMENTS: Experimental Procedure, Report template (pdf), Report template (doc), Spectra INTRODUCTION In an "aldol addition" reaction, an enol or more commonly an enolate of an aldehyde or ketone reacts with a second aldehyde or ketone forming a new carbon-carbon bond. This makes the aldol reaction an important reaction for organic synthesis. Originally, the aldol reaction used ethanal (see below) and therefore the product contained both an aldehyde and an alcohol functional group; thus it became known as the aldol reaction. Recall that aldehydes and ketones typically undergo nucleophilic addition reactions. The aldol reaction demonstrates how carbonyl compounds can react as electrophiles or nucleophiles depending on the reaction conditions and what other species are present. Alcohols are similar in that respect. Can you think of examples to illustrate that ? The aldol reaction requires an aldehyde or ketone that contains at least one -hydrogen (the -hydrogen is on the carbon adjacent to the C=O group) since the -hydrogen is required in order to form the enol or enolate. In the base-catalysed aldol reaction, the relatively acidic hydrogen on the -carbon (typical pKa 16-20) is deprotonated by a base to form the enolate. The enolate reacts as a carbon nucleophile that can then react with the electrophilic carbonyl carbon of another aldehyde or ketone molecule. Depending on the strength of the base used, the extent of deprotonation can be controlled. If a strong base is used (such as lithium diisopropylamide, LDA) then deprotonation is quantitative (100%). -

Copyrighted Material
JWST960-SUBIND JWST960-Smith October 25, 2019 9:5 Printer Name: Trim: 254mm × 178mm SUBJECT INDEX The vast use of transition metal catalysts in organic chemistry makes the citation of every individual metal impractical, so there are limited citations of individual metals. Palladium is one exception where individual citations are common, in keeping with the widespread use of that metal. However, in most cases, the term metal catalyst, or catalyst, metal is used as a heading, usually representing transition metals. A-SE2 mechanism 893 and the steering wheel model acceleration of Diels-Alder reactions 487 151–152 reactions, high pressure A1 mechanism, acetal hydrolysis and universal NMR database 1038 487 155 hydrogen-bonding 1038 A1,3-strain 196 Cahn-Ingold-Prelog system hydrophobic effect 1038 A2 mechanism, acetal hydrolysis 149–152 in water 1038 487 determination 152 ionic liquids 1038 ab initio calculations 36 D/L nomenclature 149 micellular effects 1038 and acidity 346 Kishi’s NMR method 155 microwave irradiation 1038 and antiaromaticity 71 sequence rules 149–152 phosphate 1039 and nonclassical carbocations absolute hardness 64, 359, 361 solid state 1038 427 table 361 ultracentrifuge 1038 norbornyl carbocation 436 absorbents, chiral 168 ultrasound 1038 ab initio studies 248 absorption, and conjugation 317 zeolites 1038 1,2-alkyl shifts in alkyne anions differential, and diastereomers acceleration, Petasis reaction 1349 168 1202 and cubyl carbocation 413 differential, and resolution 169 acenaphthylene, reaction with and SN2 408–409 abstraction, -

Paper : HCHE4CC09LTH
Paper : HCHE4CC09LTH Module-1: Exploitation of acidity of α-H of C=O and organometallics Mannich reaction The condensation of a CH-activated compound (usually an aldehyde or ketone) with a primary or secondary amine (or ammonia) and a nonenolizable aldehyde (or ketone) to afford aminoalkylated derivatives is known as the Mannich reaction. More generally it is The t hree-component aminomethylation from amine, aldehyde and a compound with an acidic methylene moiety. Mechanism The mechanism of the Mannich reaction has been extensively investigated. The reaction can proceed under both acidic and basic conditions, but acidic conditions are more common. Under acidic conditions the first step is the reaction of the amine component with the protonated non-enolizable carbonyl compound to give a hemiaminal, which after proton transfer loses a molecule of water to give the electrophilic iminium ion.50 This iminium ion then reacts with the enolized carbonyl compound (nucleophile) at its α-carbon in an aldol-type reaction to give rise to the Mannich base. Mannich reaction Examples Asymmetric Mannich Reaction Perkin Reaction The condensation of aromatic aldehydes with the anhydrides of aliphatic carboxylic acids in the presence of a weak base to afford α,β-unsaturated carboxylic acids is known as the Perkin reaction or Perkin condensation . The general features of the transformation are: 1) The aldehyde component is most often aromatic, but aliphatic aldehydes with no α-hydrogens as well as certain α,β-unsaturated aldehydes can also be used. 2) the reaction is more facile and gives higher yield of the product when the aromatic aldehyde has one or more electron-withdrawing substituents.