Conjugate Addition Approach for Natural Product Synthesis Inspired by Gibberellin and Solanoeclepin a Targets*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mint and Mint Oil Profile New York State Integrated Pest Management Cornell Cooperative Extension Program
http://hdl.handle.net/1813/56133 Mint and Mint Oil Profile New York State Integrated Pest Management Cornell Cooperative Extension Program Mint and Mint Oil Profile Active Ingredient Eligible for Minimum Risk Pesticide Use Brian P. Baker and Jennifer A. Grant New York State Integrated Pest Management, Cornell University, Geneva NY Label Display Names: Cornmint, Cornmint oil, U.S. EPA PC Code: 128800 (Peppermint is also Spearmint, Spearmint oil cross referenced under the listing for mint and mint oil) Active Components: Carvone, Menthol, pulegone, limonene, linarin, pinene, piperitone, linalool, and CA DPR Chem Code: Not Found various other terpenoids, alcohols, esters, and hydrocarbons Other Names: Cornmint oil, spearmint oil, men- thol oil, spearmint terpenes CAS Registry #s: 8008-79-5 (Spearmint oil) Other Codes: 68917-18-0 (Cornmint oil) Cornmint oil—FEMA: 4219; EINECS: 290-058-5; HT: None (Cornmint and Spearmint) 3301.25 Spearmint oil—FEMA: 3032; EINECS: 283-656- HT: 3301.25; RTECS: WG7360000 Summary: Mint and mint oils are a class of active ingredients derived from selected members of the plant genus Mentha. Primary sources include cornmint, peppermint, and spearmint. Active substances contained in the plants and essential oils in this class include menthol, carvone, and various other ter- penoids. Peppermint and spearmint are commonly used food ingredients; cornmint is used as a food ingredient in some Asian cuisines and is a major source of food-grade menthol used as a flavoring agent. As a pesticide, mint and mint oils work primarily through non-toxic modes of action as repellents, but also have anti-microbial properties. Peppermint is covered in a separate profile. -

Proton Nmr Spectroscopy
1H NMR Spectroscopy (#1c) The technique of 1H NMR spectroscopy is central to organic chemistry and other fields involving analysis of organic chemicals, such as forensics and environmental science. It is based on the same principle as magnetic resonance imaging (MRI). This laboratory exercise reviews the principles of interpreting 1H NMR spectra that you should be learning right now in Chemistry 302. There are four questions you should ask when you are trying to interpret an NMR spectrum. Each of these will be discussed in detail. The Four Questions to Ask While Interpreting Spectra 1. How many different environments are there? The number of peaks or resonances (signals) in the spectrum indicates the number of nonequivalent protons in a molecule. Chemically equivalent protons (magnetically equivalent protons) give the same signal in the NMR whereas nonequivalent protons give different signals. 1 For example, the compounds CH3CH3 and BrCH2CH2Br all have one peak in their H NMR spectra because all of the protons in each molecule are equivalent. The compound below, 1,2- dibromo-2-methylpropane, has two peaks: one at 1.87 ppm (the equivalent CH3’s) and the other at 3.86 ppm (the CH2). 1.87 1.87 ppm CH 3 3.86 ppm Br Br CH3 1.87 ppm 3.86 10 9 8 7 6 5 4 3 2 1 0 2. How many 1H are in each environment? The relative intensities of the signals indicate the numbers of protons that are responsible for individual signals. The area under each peak is measured in the form of an integral line. -

SAFETY DATA SHEET Isopropyl Alcohol
SAFETY DATA SHEET Isopropyl Alcohol Section 1. Identification GHS product identifier : Isopropyl Alcohol Chemical name : Isopropyl alcohol Other means of : isopropanol; 2-Propanol identification Product type : Liquid. Product use : Synthetic/Analytical chemistry. Synonym : isopropanol; 2-Propanol SDS # : 001105 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 2 substance or mixture EYE IRRITATION - Category 2A SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : May form explosive mixtures with air. Highly flammable liquid and vapor. Causes serious eye irritation. May cause drowsiness or dizziness. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Prevention : Wear protective gloves. Wear eye or face protection. Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. Use explosion- proof electrical, ventilating, lighting and all material-handling equipment. Use only non- sparking tools. Take precautionary measures against static discharge. Keep container tightly closed. Use only outdoors or in a well-ventilated area. Avoid breathing vapor. Wash hands thoroughly after handling. Response : IF INHALED: Remove person to fresh air and keep comfortable for breathing. Call a POISON CENTER or physician if you feel unwell. -

There Are Three Major Biological Molecules Classified As Ketone Bodies
There are three major biological molecules classified as ketone bodies: These ketone bodies are water soluble and do not need specific transporters to cross membranes. Synthesis of acetoacetate 1. React two acetyl-CoA molecules with each other using thiolase. This is called acetoacetyl-CoA. What is the second product? 2. React a third acetyl-CoA molecule with acetoacetyl-CoA. This step is catalyzed by hydroxymethylglutaryl-CoA synthase (HMG-CoA synthase). a. Deprotonate C2 of acetyl-CoA. You have created a great nucleophile. b. React your newly formed carbanion nucleophile with the electrophilic carbonyl C3 of acetoacetyl-CoA. c. Protonate the oxyanion. d. Use water as a nucleophile to react with the electrophilic carbonyl of the thioester of the newly added acetyl-CoA unit. This results in a carboxylate functional group. e. Your product should contain a 5-carbon chain, which starts with a thioester to CoA, ends with a carboxylate, and has a hydroxyl and a methyl group attached to C3. This is β-hydroxy-β- methylglutaryl-CoA (HMG-CoA). 3. An acetyl-CoA group is eliminated. This step is catalyzed by hydroxymethylglutaryl-CoA lyase (HMG- CoA lyase). a. A base deprotonates the hydroxyl group of β-hydroxy-β-methylglutaryl-CoA. b. A pair of electrons from the oxyanion moves to form a carbonyl. C2 leaves as a carbanion [which delocalizes into the adjacent thioester carbonyl]. c. The first product is acetoacetate. d. The carbanion picks up the proton and leaves as acetyl-CoA. Formation of acetone from acetoacetate This occurs in a non-enzymatic fashion because of the arrangement of the ketone in the β position from the carboxylate in acetoacetate and causes problems since acetone builds up. -

DEVELOPMENT of NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS by LINDSEY RAE CULLEN DISSERTATION Submitted
DEVELOPMENT OF NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS BY LINDSEY RAE CULLEN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Scott E. Denmark, Chair Professor Martin D. Burke Professor Scott K. Silverman Professor Steven C. Zimmerman ii Abstract The first half of this thesis (Chapter 2) described the development of a fluoride-promoted conjugate addition of sulfur-stabilized carbanion nucleophiles to α,β-unsaturated ketones and esters. This reaction was achieved using a substoichiometric amount of TBAF, resulting in high yields on the desired 1,4-addition product. The addition of 1,3-dithianes was given particular focus as a novel method for the preparation of differentially protect 1,4-dicarbonyl compounds. Observation by 13C NMR spectroscopy provided evidence that the reaction proceeds through an ion pair, and attempts to extend this reaction to asymmetric additions using a chiral counterion are presented in detail. The second half of this thesis (Chapter 3) details development of a phase transfer catalyzed [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds. Initial investigation focused on identifying viable substrate classes that would undergo selective [2,3]-rearrangement under phase transfer conditions. Under certain conditions, the [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds takes place in the presence of a phase transfer agent, providing a rare example of a phase transfer catalyzed unimolecular reaction. In the course of this investigation it was found that catalysis is dependent on several variables including base concentration, catalyst structure, and substrate lipophilicity. -
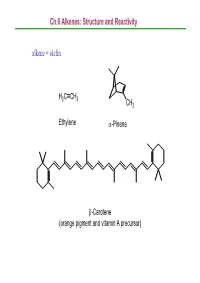
Ch.6 Alkenes: Structure and Reactivity Alkene = Olefin
Ch.6 Alkenes: Structure and Reactivity alkene = olefin H2CCH2 CH3 Ethylene α-Pinene β-Carotene (orange pigment and vitamin A precursor) Ch.6 Alkenes: Structure and Reactivity 6.1 Industrial Preparation and Use of Alkenes Compounds derived industrially from ethylene CH3CH2OH Ethanol CH3CHO Acetaldehyde CH3COOH Acetic acid HOCH2CH2OH Ethylene glycol ClCH2CH2Cl Ethylene dichloride H C=CHCl Vinyl chloride H2CCH2 2 O Ethylene oxide Ethylene (26 million tons / yr) O Vinyl acetate O Polyethylene Ch.6 Alkenes: Structure and Reactivity Compounds derived industrially from propylene OH Isopropyl alcohol H3CCH3 O Propylene oxide CH3 H3CCH CH2 Propylene Cumene (14 million tons / yr) CH3 CH3 Polypropylene Ch.6 Alkenes: Structure and Reactivity • Ethylene, propylene, and butene are synthesized industrially by thermal cracking of natural gas (C1-C4 alkanes) and straight-run gasoline (C4-C8 alkanes). 850-900oC CH (CH ) CH H + CH + H C=CH + CH CH=CH 3 2 n 3 steam 2 4 2 2 3 2 + CH3CH2CH=CH2 - the exact processes are complex; involve radical process H 900oC CH3CH2 CH2CH3 22H2CCH H2C=CH2 +H2 Ch.6 Alkenes: Structure and Reactivity • Thermal cracking is an example of a reaction whose energetics are dominated by entropy (∆So) rather than enthalpy (∆Ho) in the free-energy equation (∆Go = ∆Ho -T∆So) . ; C-C bond cleavage (positive ∆Ho) ; high T and increased number of molecules → larger T∆So Ch.6 Alkenes: Structure and Reactivity 6.2 Calculating Degree of Unsaturation unsaturated: formula of alkene CnH2n ; formula of alkane CnH2n+2 in general, each ring or double -

N-BUTYL ALCOHOL
Right to Know Hazardous Substance Fact Sheet Common Name: n-BUTYL ALCOHOL Synonyms: Propyl Carbinol; n-Butanol CAS Number: 71-36-3 Chemical Name: 1-Butanol RTK Substance Number: 1330 Date: November 1998 Revision: January 2008 DOT Number: UN 1120 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE n-Butyl Alcohol is a colorless liquid with a strong, sweet Hazard Summary alcohol odor. It is used as a solvent for fats, waxes, shellacs, Hazard Rating NJDOH NFPA resins, gums, and varnish, in making hydraulic fluids, and in HEALTH - 2 medications for animals. FLAMMABILITY - 3 REACTIVITY - 0 f ODOR THRESHOLD = 1 to 15 ppm FLAMMABLE f Odor thresholds vary greatly. Do not rely on odor alone to POISONOUS GASES ARE PRODUCED IN FIRE determine potentially hazardous exposures. CONTAINERS MAY EXPLODE IN FIRE Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; Reasons for Citation 4=severe f n-Butyl Alcohol is on the Right to Know Hazardous f n-Butyl Alcohol can affect you when inhaled and by Substance List because it is cited by OSHA, ACGIH, DOT, passing through the skin. NIOSH, DEP, IRIS, NFPA and EPA. f Contact can irritate and burn the skin. f This chemical is on the Special Health Hazard Substance f n-Butyl Alcohol can irritate and burn the eyes with possible List. eye damage. f Inhaling n-Butyl Alcohol can irritate the nose, throat and lungs. f Exposure to n-Butyl Alcohol can cause headache, dizziness, nausea and vomiting. SEE GLOSSARY ON PAGE 5. f n-Butyl Alcohol can damage the liver, kidneys, hearing, and sense of balance. -
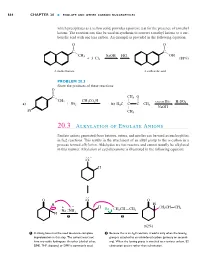
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Ganic Compounds
6-1 SECTION 6 NOMENCLATURE AND STRUCTURE OF ORGANIC COMPOUNDS Many organic compounds have common names which have arisen historically, or have been given to them when the compound has been isolated from a natural product or first synthesised. As there are so many organic compounds chemists have developed rules for naming a compound systematically, so that it structure can be deduced from its name. This section introduces this systematic nomenclature, and the ways the structure of organic compounds can be depicted more simply than by full Lewis structures. The language is based on Latin, Greek and German in addition to English, so a classical education is beneficial for chemists! Greek and Latin prefixes play an important role in nomenclature: Greek Latin ½ hemi semi 1 mono uni 1½ sesqui 2 di bi 3 tri ter 4 tetra quadri 5 penta quinque 6 hexa sexi 7 hepta septi 8 octa octo 9 ennea nona 10 deca deci Organic compounds: Compounds containing the element carbon [e.g. methane, butanol]. (CO, CO2 and carbonates are classified as inorganic.) See page 1-4. Special characteristics of many organic compounds are chains or rings of carbon atoms bonded together, which provides the basis for naming, and the presence of many carbon- hydrogen bonds. The valency of carbon in organic compounds is 4. Hydrocarbons: Compounds containing only the elements C and H. Straight chain hydrocarbons are named according to the number of carbon atoms: CH4, methane; C2H6 or H3C-CH3, ethane; C3H8 or H3C-CH2-CH3, propane; C4H10 or H3C-CH2- CH2-CH3, butane; C5H12 or CH3CH2CH2CH2CH3, pentane; C6H14 or CH3(CH2)4CH3, hexane; C7H16, heptane; C8H18, octane; C9H20, nonane; C10H22, CH3(CH2)8CH3, decane. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Williamson Ether Synthesis the Williamson Ether Synthesis Is an Organic Reaction, Forming an Ether from an Alkyl Halide and an Alcohol
The Williamson ether synthesis The Williamson ether synthesis is an organic reaction, forming an ether from an alkyl halide and an alcohol. This reaction was developed by Alexander Williamson in 1850. It involves the reaction of an alkoxide ion with a primary alkyl halide via an SN2 reaction. The Williamson reaction is widely used in both laboratory and industrial synthesis, and remains the simplest and most popular method of preparing ethers. Both symmetrical and asymmetrical ethers are easily prepared. The reaction for this week: an example of a Williamson ether synthesis acetaminophen ethyl iodide phenacetin starting material reagent product Phenacetin may be synthesized as an example of the Williamson ether synthesis The first synthesis of phenacetin was reported in 1878 by Harmon Morse. Procedure 1. Weigh an Extra-Strength Tylenol tablet. Pulverize the tablet with mortar and pestle. Weigh out 0.22 g and place it in a dry 15-ml round-bottom flask along with 0.28 g of finely pulverized K2CO3 (mortar and pestle) and 3.0 mL of butanone. Carefully add 0.28 mL of ethyl iodide with a syringe. 2. Add a stir bar; attach a microscale water-cooled condenser to the flask. Heat the mixture under reflux directly on a hot plate at medium setting for 1 hour. In the meantime, obtain the IR of acetaminophen. 3. Turn off the heat. Allow the mixture to cool down. Add 4 mL of water to the flask and transfer its contents to a 16 x 125 mm test tube with a screw cap. Rinse round-bottom flask 4 times with 1 mL of tert-butyl methyl ether (BME) and add the rinsings to the test tube. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3.