Carbonyl Condensation Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hereditary Galactokinase Deficiency J
Arch Dis Child: first published as 10.1136/adc.46.248.465 on 1 August 1971. Downloaded from Alrchives of Disease in Childhood, 1971, 46, 465. Hereditary Galactokinase Deficiency J. G. H. COOK, N. A. DON, and TREVOR P. MANN From the Royal Alexandra Hospital for Sick Children, Brighton, Sussex Cook, J. G. H., Don, N. A., and Mann, T. P. (1971). Archives of Disease in Childhood, 46, 465. Hereditary galactokinase deficiency. A baby with galactokinase deficiency, a recessive inborn error of galactose metabolism, is des- cribed. The case is exceptional in that there was no evidence of gypsy blood in the family concerned. The investigation of neonatal hyperbilirubinaemia led to the discovery of galactosuria. As noted by others, the paucity of presenting features makes early diagnosis difficult, and detection by biochemical screening seems desirable. Cataract formation, of early onset, appears to be the only severe persisting complication and may be due to the biosynthesis and accumulation of galactitol in the lens. Ophthalmic surgeons need to be aware of this enzyme defect, because with early diagnosis and dietary treatment these lens changes should be reversible. Galactokinase catalyses the conversion of galac- and galactose diabetes had been made in this tose to galactose-l-phosphate, the first of three patient (Fanconi, 1933). In adulthood he was steps in the pathway by which galactose is converted found to have glycosuria as well as galactosuria, and copyright. to glucose (Fig.). an unexpectedly high level of urinary galactitol was detected. He was of average intelligence, and his handicaps, apart from poor vision, appeared to be (1) Galactose Gackinase Galactose-I-phosphate due to neurofibromatosis. -

Alkyl Halides Work Best; Secondary Alkyl Halides Give Lower Yields
CHAPTER 21 ESTER ENOLATES ou have already had considerable experience with carbanionic compounds and their applications in synthetic organic chemistry. The first was acetylide ion in YChapter 9, followed in Chapter 14 by organometallic compounds—Grignard reagents, for example—that act as sources of negatively polarized carbon. In Chapter 18 you learned that enolate ions—reactive intermediates generated from aldehydes and ketones—are nucleophilic, and that this property can be used to advantage as a method for carbon–carbon bond formation. The present chapter extends our study of carbanions to the enolate ions derived from esters. Ester enolates are important reagents in synthetic organic chemistry. The stabilized enolates derived from -keto esters are particularly useful. OO C ␣ C  R CH2 ORЈ -Keto ester: a ketone carbonyl is  to the carbonyl group of the ester. A proton attached to the ␣-carbon atom of a -keto ester is relatively acidic. Typical Ϫ11 acid dissociation constants Ka for -keto esters are Ϸ10 (pKa 11). Because the ␣- carbon atom is flanked by two electron-withdrawing carbonyl groups, a carbanion formed at this site is highly stabilized. The electron delocalization in the anion of a -keto ester is represented by the resonance structures 831 832 CHAPTER TWENTY-ONE Ester Enolates Ϫ Ϫ O O O O O O C C C C C C R C ORЈ R ϪC ORЈ R C ORЈ H H H Principal resonance structures of the anion of a -keto ester We’ll begin by describing the preparation and properties of -keto esters, proceed to a discussion of their synthetic applications, continue to an examination of related species, and conclude by exploring some recent developments in the active field of synthetic car- banion chemistry. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

Enantioselective Solvent-Free Robinson Annulation Reactions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Publications of the IAS Fellows Proc. Indian Acad. Sci. (Chem. Sci.), Vol. 113, No. 3, June 2001, pp 197–213 Ó Indian Academy of Sciences Enantioselective solvent-free Robinson annulation reactions D RAJAGOPAL, R NARAYANAN and S SWAMINATHAN* Department of Organic Chemistry, University of Madras, Guindy Campus, Chennai 600 025, India e-mail: [email protected] MS received 28 March 2001 Abstract. The enantioselective cyclization of the prochiral cyclic substrates 1 to 7 and 26, can be carried out in the neat using S-proline as catalyst. The substrates 18 to 22 and 27 could not be cyclized with S-proline but could be cyclized with a mixture of S-phenylalanine and d-camphorsulphonic acid. The enantioselective cyclization of prochiral acyclic triones 45 and 47 and also the racemic tricarbonyl compounds 54 to 57 could also be carried out in the neat using S-proline as catalyst. The optically active enediones obtained in the above cyclizations could also be obtained directly from 1,3-diones or 2-hydroxymethylene cycloalkanones in a one-pot reaction with methyl vinyl ketone (MVK) and S-proline in the absence of solvents. 13C NMR studies of the one-pot synthesis of S-11 and S-14 reveal that the annulations involve initial formation of an acid-base complex followed by a Michael reaction and then an enantioselective cyclization. Such enantioselective cyclizations probably occur on the surface of S-proline crystals. Keywords. Enantioselective annulation; cyclization; S-proline; S-phenylalanine; d-camphorsulphonic acid. -

Robinson Annulation
Robinson annulation The Robinson annulation is a chemical reaction used in organic Robinson annulation chemistry for ring formation. It was discovered by Robert Robinson Named after Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds.[1] The method uses a ketone and a methyl Reaction type Ring forming vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring reaction by a Michael addition followed by an aldol condensation. This Identifiers procedure is one of the key methods to form fused ring systems. Organic robinson-annulation Chemistry Portal RSC ontology RXNO:0000380 ID Formation of cyclohexenone and derivatives are important in chemistry for their application to the synthesis of many natural products and other interesting organic compounds such as antibiotics and steroids.[2] Specifically, the synthesis of cortisone is completed through the use of the Robinson annulation.[3] The initial paper on the Robinson annulation was published by William Rapson and Robert Robinson while Rapson studied at Oxford with professor Robinson. Before their work, cyclohexenone syntheses were not derived from the α,β-unsaturated ketone component. Initial approaches coupled the methyl vinyl ketone with a naphthol to give a naphtholoxide, but this procedure was not sufficient to form the desired cyclohexenone. This was attributed to unsuitable conditions of the reaction.[1] Robinson and Rapson found in 1935 that the interaction between cyclohexanone and α,β-unsaturated ketone afforded the -

In Silico Screening of Sugar Alcohol Compounds to Inhibit Viral Matrix Protein VP40 of Ebola Virus
Molecular Biology Reports (2019) 46:3315–3324 https://doi.org/10.1007/s11033-019-04792-w ORIGINAL ARTICLE In silico screening of sugar alcohol compounds to inhibit viral matrix protein VP40 of Ebola virus Nagasundaram Nagarajan1 · Edward K. Y. Yapp2 · Nguyen Quoc Khanh Le1 · Hui‑Yuan Yeh1 Received: 28 December 2018 / Accepted: 28 March 2019 / Published online: 13 April 2019 © Springer Nature B.V. 2019 Abstract Ebola virus is a virulent pathogen that causes highly lethal hemorrhagic fever in human and non-human species. The rapid growth of this virus infection has made the scenario increasingly complicated to control the disease. Receptor viral matrix protein (VP40) is highly responsible for the replication and budding of progeny virus. The binding of RNA to VP40 could be the crucial factor for the successful lifecycle of the Ebola virus. In this study, we aimed to identify the potential drug that could inhibit VP40. Sugar alcohols were enrich with antiviral properties used to inhibit VP40. Virtual screening analysis was perform for the 48 sugar alcohol compounds, of which the following three compounds show the best binding afnity: Sorbitol, Mannitol and Galactitol. To understand the perfect binding orientation and the strength of non-bonded interactions, individual molecular docking studies were perform for the best hits. Further molecular dynamics studies were conduct to analyze the efcacy between the protein–ligand complexes and it was identify that Sorbitol obtains the highest efcacy. The best-screened compounds obtained drug-like property and were less toxic, which could be use as a potential lead compound to develop anti-Ebola drugs. -
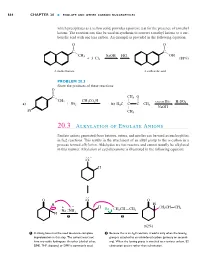
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Hemicellulose Arabinogalactan Hydrolytic Hydrogenation Over Ru-Modified H-USY Zeolites
Research Collection Journal Article Hemicellulose arabinogalactan hydrolytic hydrogenation over Ru-modified H-USY zeolites Author(s): Murzin, Dmitry; Kusema, Bright; Murzina, Elena V.; Aho, Atte; Tokarev, Anton; Boymirzaev, Azamat S.; Wärnå, Johan; Dapsens, Pierre Y.; Mondelli, Cecilia; Pérez-Ramírez, Javier; Salmi, Tapio Publication Date: 2015-10 Permanent Link: https://doi.org/10.3929/ethz-a-010792434 Originally published in: Journal of Catalysis 330, http://doi.org/10.1016/j.jcat.2015.06.022 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library Hemicellulose arabinogalactan hydrolytic hydrogenation over Ru-modified H-USY zeolites Dmitry Yu. Murzin1*, Bright Kusema2, Elena V. Murzina1, Atte Aho1, Anton Tokarev1, Azamat S. Boymirzaev3, Johan Wärnå1,4, Pierre Y. Dapsens2, Cecilia Mondelli2, Javier Pérez-Ramírez2, Tapio Salmi1 1Laboratory of Industrial Chemistry and Reaction Engineering, Process Chemistry Centre, Department of Chemical Engineering, Åbo Akademi University, FI-20500 Åbo/Turku, Finland, E-mail: [email protected] 2Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, CH-8093 Zurich, Switzerland 3Namangan Institute of Engineering and Technology, Department of Chemical Technology, Namangan, 160115, Uzbekistan 4University of Umeå, Umeä, Sweden ABSTRACT The hydrolytic hydrogenation of hemicellulose arabinogalactan was investigated in the presence of protonic and Ru (1-5 wt.%)-modified USY zeolites (Si/Al ratio = 15 and 30). The use of the purely acidic materials was effective in depolymerizing the macromolecule into free sugars. While the latter partly dehydrated into 5- hydroxymethylfurfural and furfural, the generation of high molecular-weight compounds (aggregates of sugars and humins) was not favored, in contrast to previous evidences over beta zeolites. -

Sugar Alcohols a Sugar Alcohol Is a Kind of Alcohol Prepared from Sugars
Sweeteners, Good, Bad, or Something even Worse. (Part 8) These are Low calorie sweeteners - not non-calorie sweeteners Sugar Alcohols A sugar alcohol is a kind of alcohol prepared from sugars. These organic compounds are a class of polyols, also called polyhydric alcohol, polyalcohol, or glycitol. They are white, water-soluble solids that occur naturally and are used widely in the food industry as thickeners and sweeteners. In commercial foodstuffs, sugar alcohols are commonly used in place of table sugar (sucrose), often in combination with high intensity artificial sweeteners to counter the low sweetness of the sugar alcohols. Unlike sugars, sugar alcohols do not contribute to the formation of tooth cavities. Common Sugar Alcohols Arabitol, Erythritol, Ethylene glycol, Fucitol, Galactitol, Glycerol, Hydrogenated Starch – Hydrolysate (HSH), Iditol, Inositol, Isomalt, Lactitol, Maltitol, Maltotetraitol, Maltotriitol, Mannitol, Methanol, Polyglycitol, Polydextrose, Ribitol, Sorbitol, Threitol, Volemitol, Xylitol, Of these, xylitol is perhaps the most popular due to its similarity to sucrose in visual appearance and sweetness. Sugar alcohols do not contribute to tooth decay. However, consumption of sugar alcohols does affect blood sugar levels, although less than that of "regular" sugar (sucrose). Sugar alcohols may also cause bloating and diarrhea when consumed in excessive amounts. Erythritol Also labeled as: Sugar alcohol Zerose ZSweet Erythritol is a sugar alcohol (or polyol) that has been approved for use as a food additive in the United States and throughout much of the world. It was discovered in 1848 by British chemist John Stenhouse. It occurs naturally in some fruits and fermented foods. At the industrial level, it is produced from glucose by fermentation with a yeast, Moniliella pollinis. -

Oleaginous Yeasts for the Production of Sugar Alcohols Sujit S Jagtap1,2
Oleaginous Yeasts for the Production of Sugar Alcohols Sujit S Jagtap1,2* ([email protected]), Ashwini A Bedekar2, Jing-Jing Liu1, Anshu Deewan1,2, and Christopher V Rao1,2 1DOE Center for Advanced Bioenergy and Bioproducts Innovation, University of Illinois at Urbana-Champaign, Illinois. 2Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Illinois. https://cabbi.bio/ Project Goal: The goal of this project is to investigate sugar alcohol production from plant- based sugars and glycerol in the oleaginous yeasts Rhodosporidium toruloides and Yarrowia lipolytica. We are also interested in understanding the mechanism of sugar alcohol production and the key genes involved in the polyol synthesis process. Sugar alcohols are commonly used as low-calorie, natural sweeteners. They have also been proposed by the Department of Energy as potential building blocks for bio-based chemicals. They can be used to produce polymers with applications in medicine and as precursors to anti-cancer drugs 1. Production of these sugar alcohols by yeast often results, from redox imbalances associated with growth on different sugars, accumulation of toxic intermediates, and as a cell response to the high osmotic pressure of the environment 2-3. The ability of yeast to naturally produce these sugar alcohols from simple sugars provides a potentially safer and more sustainable alternative to traditional chemical hydrogenation. In our study, we found that the oleaginous yeast R. toruloides IFO0880 produces D-arabitol during growth on xylose in nitrogen-rich medium 3. Efficient xylose utilization was a prerequisite for extracellular D-arabitol production. D-arabitol is an overflow metabolite associated with transient redox imbalances during growth on xylose. -

Cross-Coupling Reactions Using Samarium(II) Iodide Michal Szostak,* Neal J
Review pubs.acs.org/CR Cross-Coupling Reactions Using Samarium(II) Iodide Michal Szostak,* Neal J. Fazakerley, Dixit Parmar, and David J. Procter* School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, United Kingdom 4.3. Aldol Reactions BM 5. Cross-Coupling As Part of Sequential and Cascade Reactions BM 5.1. Cascades Initiated by Radical Intermediates BN 5.2. Cascades Initiated by Anionic Intermediates BR 6. Conclusions BT Author Information BT Corresponding Authors BT Notes BT Biographies BT CONTENTS Acknowledgments BU 1. Introduction A References BU 2. Reactivity of Functional Groups toward Samarium Note Added in Proof CB Diiodide B 3. Cross-Coupling via Radical Intermediates C 3.1. Ketyl Radical-Alkene/Alkyne/Arene Cross- 1. INTRODUCTION Coupling C Since its introduction to organic synthesis in 1977 by Kagan,1,2 ’ 3.1.1. Intramolecular Cross-Coupling of Ketyl samarium(II) iodide (SmI2, Kagan s reagent) has gained the Radicals with Alkenes C status of one of the most versatile single-electron transfer − 3.1.2. Intermolecular Cross-Coupling of Ketyl reagents available in the laboratory.3 46 SmI occupies a unique − 2 Radicals with Alkenes S place among other reductants47 57 in that it is an extremely − 3.1.3. Cross-Coupling of Ketyl Radicals with powerful58 64 yet chemoselective reagent, whose selectivity Alkynes Y toward functional groups is fine-tuned by the use of appropriate 26−30 3.1.4. Cross-Coupling of Ketyl Radicals with ligands and additives. Transformations mediated by SmI2 Allenes Z are performed under user-friendly and operationally simple 3.1.5. -

Retinal Polyol and Myo-Lnosifol in Galactosemic Dogs Given an Aldose-Reducto.Se Inhibitor
Investigative Ophthalmology & Visual Science, Vol. 32, No. 13, December 1991 Copyright © Association for Research in Vision and Ophthalmology Retinal Polyol and Myo-lnosifol in Galactosemic Dogs Given an Aldose-Reducto.se Inhibitor Timothy 5. Kern and Ronald L. Engerman Galactitol and myo-\nosito\ concentrations were measured in retinas, erythrocytes, and skeletal muscle of experimentally galactosemic dogs receiving a placebo or the aldose reductase inhibitor, sorbinil, for 5 yr. The concentration of galactitol was increased more than 30-fold in the retina and other tissues by galactosemia, and the increase was inhibited 90-96% in all tissues by sorbinil. The concentration of free myo-inos\to\ was greater than normal in retinas of galactosemic dogs, and its concentration was not altered by the aldose-reductase inhibitor. The mjw-inositol concentration likewise was greater than normal in the retinas of dogs that were diabetic for 2-4 months. The marked inhibition of polyol production and accumulation in the retina of sorbinil-treated galactosemic dogs was not associated with a comparable inhibition of retinopathy. Invest Ophthalmol Vis Sci 32:3175-3177,1991 Experimental elevation of blood galactose concen- to receive either the aldose reductase inhibitor, sor- tration in normal dogs leads to a retinopathy that is binil, or to remain untreated. The dogs were fed twice morphologically similar to that in diabetic dogs and daily (8 AM and 6 PM) to maintain blood galactose humans.12 One mechanism by which hyperglycemia levels elevated as high as possible throughout the day. in diabetes or experimental galactosemia might cause Sorbinil was given orally twice a day, usually at a dose retinopathy is through excessive polyol production of 60-80 mg/kg/day, 30 min before each feeding.