Alkyl Halides Work Best; Secondary Alkyl Halides Give Lower Yields
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 1 Tropone and Tropolone
School of Molecular and Life Sciences New Routes to Troponoid Natural Products Jason Matthew Wells This thesis is presented for the Degree of Doctor of Philosophy of Curtin University November 2018 Declaration To the best of my knowledge and belief this thesis contains no material previously pub- lished by any other person except where due acknowledgement has been made. This thesis contains no material which has been accepted for the award of any other degree or diploma in any other university. Signature: Date: i Abstract Malaria is an infectious disease found in humans and other animals, it is caused by a single-cell parasite of the Plasmodium genus with many different substrains. Of these, P. falciparum is the most deadly to humans causing the majority of deaths. Although research into the area of antimalarial compounds is wide spread, few have been devel- oped with new structural features. Cordytropolone 37 is a natural product isolated in 2001 from the insect pathogenic fungus Cordyceps sp. BCC 1681 and has been shown to have antimalarial activity against P. falciparum. It has a structure unrelated to antimalarial com- pounds currently used in therapy. It does not contain a peroxide bridge as with artemisinin 25 or quinoline rings as with chloroquine 22. This unique structure indicates that it could possibly interact with the malaria parasite in a fashion unlike current treatments. In order for cordytropolone to be further developed as a potential treatment, it must first be synthe- sised in a laboratory environment. This study attempts to develop the first total synthesis of cordytropolone. H HO O N O O N O N H H H O O Cl HO O 22 25 37 Figure 0.0.1: Cordytropolone 37 has a unique structure compared to the current common malaria treatments The first method investigated towards the total synthesis of cordytropolone involved an intramolecular Buchner ring expansion. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

Landolt-Börnstein Indexes of Organic Compounds Subvolumes A-I by V
Landolt-Börnstein Indexes of Organic Compounds Subvolumes A-I By V. Vill, C. Bauhofer, G. Peters, H. Sajus, P. Weigner, LCI-Publisher and Chemistry Department of the University of Hamburg All printed index material has been used to build up the comprehensive Scidex database index developed by LCI Publisher GmbH, Hamburg For further information please visit www.lci-publisher.com From this database a CD-ROM and two online versions were derived. The first is attached to each of the printed subvolumes and the latter are offered for free use at the following addresses: Scidex Database online with graphical structure search on http://lb.chemie.uni-hamburg.de/ Or the easy to use html version on http://lb.chemie.uni-hamburg.de/static/ Landolt-Börnstein Numerical Data and Functional Relationships in Science and Technology New Series / Editor in Chief: W. Martienssen Index of Organic Compounds Subvolume A Compounds with 1 to 7 Carbon Atoms Editor: V. Vill Authors: V. Vill, G. Peters, H. Sajus 1 3 ISBN 3-540-66203-0 Springer-Verlag Berlin Heidelberg New York Library of Congress Cataloging in Publication Data Zahlenwerte und Funktionen aus Naturwissenschaften und Technik, Neue Serie Editor in Chief: W. Martienssen Index of Organic Compounds A: Editor: V. Vill At head of title: Landolt-Börnstein. Added t.p.: Numerical data and functional relationships in science and technology. Tables chiefly in English. Intended to supersede the Physikalisch-chemische Tabellen by H. Landolt and R. Börnstein of which the 6th ed. began publication in 1950 under title: Zahlenwerte und Funktionen aus Physik, Chemie, Astronomie, Geophysik und Technik. -
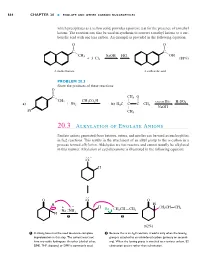
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Carboxylic Acids and Their Derivatives
8 CARBOXY A lmost all of the basic types of reactions now have been covered: addition, elimination, substitution, and rearrangement by polar, radical, and concerted mechanisms. Indeed, if you have been looking for similarities, you will have seen that most of the reactions discussed in the preceding three chapters are variations on basic types we have discussed earlier. Furthermore, most of the basic structural effects that determine chemical reactivity also have been covered in previous chapters: bond energies, steric hindrance, electronega- tivity, electron delocalization, hydrogen bonding, solvation, and conforma- tional influences. You might well ask what is left. The answer is, a great deal- but now we will be concerned mostly with putting concepts together, moving from the simple to the complex. For example, in this chapter we will be trying to under- stand the ways that carboxylic acids, which possess the -c' functional \ group, are similar to and different from alcohols, which have the OH group, and aldehydes and ketones, which have C=O bonds. Subsequently we will look at acids that also possess OH or NH, sub- stituent groups (or both) and develop a rationale for the behavior of these combinations in terms of effects we already have discussed. Insofar as pos- sible, you should try to do this yourself whenever you encounter a substance with a new set of combinations of functional groups on its molecules. You often will be in error (as many experts will be), because even if you take Carboxylic Acids and Their Derivatives 789 account of all of the structural effects, as well as the possible reactions or inter- actions, the overall result of these frequently is very difficult to judge in advance. -

Carbonyl Condensation Reactions
24 Carbonyl Condensation Reactions 24.1 The aldol reaction 24.2 Crossed aldol reactions 24.3 Directed aldol reactions 24.4 Intramolecular aldol reactions 24.5 The Claisen reaction 24.6 The crossed Claisen and related reactions 24.7 The Dieckmann reaction 24.8 The Michael reaction 24.9 The Robinson annulation Ibuprofen is the generic name for the pain reliever known by the trade names of Motrin and Advil. Like aspirin, ibuprofen acts as an anti-infl ammatory agent by blocking the synthesis of prostaglandins from arachidonic acid. One step in a commercial synthesis of ibuprofen involves the reaction of a nucleophilic enolate with an electrophilic carbonyl group. In Chap- ter 24, we learn about the carbon–carbon bond-forming reactions of enolates with carbonyl electrophiles. 916 smi75625_ch24_916-948.indd 916 11/12/09 12:12:52 PM 24.1 The Aldol Reaction 917 In Chapter 24, we examine carbonyl condensations—that is, reactions between two car- bonyl compounds—a second type of reaction that occurs at the α carbon of a carbonyl group. Much of what is presented in Chapter 24 applies principles you have already learned. Many of the reactions may look more complicated than those in previous chapters, but they are fundamen- tally the same. Nucleophiles attack electrophilic carbonyl groups to form the products of nucleo- philic addition or substitution, depending on the structure of the carbonyl starting material. Every reaction in Chapter 24 forms a new carbon–carbon bond at the ` carbon to a carbonyl group, so these reactions are extremely useful in the synthesis of complex natural products. -

Copyrighted Material
JWST960-SUBIND JWST960-Smith October 25, 2019 9:5 Printer Name: Trim: 254mm × 178mm SUBJECT INDEX The vast use of transition metal catalysts in organic chemistry makes the citation of every individual metal impractical, so there are limited citations of individual metals. Palladium is one exception where individual citations are common, in keeping with the widespread use of that metal. However, in most cases, the term metal catalyst, or catalyst, metal is used as a heading, usually representing transition metals. A-SE2 mechanism 893 and the steering wheel model acceleration of Diels-Alder reactions 487 151–152 reactions, high pressure A1 mechanism, acetal hydrolysis and universal NMR database 1038 487 155 hydrogen-bonding 1038 A1,3-strain 196 Cahn-Ingold-Prelog system hydrophobic effect 1038 A2 mechanism, acetal hydrolysis 149–152 in water 1038 487 determination 152 ionic liquids 1038 ab initio calculations 36 D/L nomenclature 149 micellular effects 1038 and acidity 346 Kishi’s NMR method 155 microwave irradiation 1038 and antiaromaticity 71 sequence rules 149–152 phosphate 1039 and nonclassical carbocations absolute hardness 64, 359, 361 solid state 1038 427 table 361 ultracentrifuge 1038 norbornyl carbocation 436 absorbents, chiral 168 ultrasound 1038 ab initio studies 248 absorption, and conjugation 317 zeolites 1038 1,2-alkyl shifts in alkyne anions differential, and diastereomers acceleration, Petasis reaction 1349 168 1202 and cubyl carbocation 413 differential, and resolution 169 acenaphthylene, reaction with and SN2 408–409 abstraction, -

University Microfilms
INFORMATION TO USERS This dissertation was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. You will find a good image of the page in the adjacent frame. 3. When a map, drawing or chart, etc., was part of the material being photographed the photographer followed a definite method in "sectioning" the material. It is customary to begin photoing at the upper left hand corner of a large sheet and to continue photoing from left to right in equal sections with a small overlap. If necessary, sectioning is continued again — beginning below the first row and continuing on until complete. 4. The majority of users indicate that the textual content is of greatest value, however, a somewhat higher quality reproduction could be made from "photographs" if essential to the understanding of the dissertation. -

Working with Hazardous Chemicals
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Oxidation of Cyclic Ketones to Dicarboxylic Acids
102 Pol. J. Chem. Tech., Vol. 20, No. Polish4, 2018 Journal of Chemical Technology, 20, 4, 102—107, 10.2478/pjct-2018-0061 Oxidation of cyclic ketones to dicarboxylic acids Dawid Lisicki, Beata Orlińska* Silesian University of Technology, Faculty of Chemistry, Department of Chemical Organic Technology and Petrochemistry, Krzywoustego 4, 44-100 Gliwice, Poland *Corresponding author: e-mail: [email protected] This paper reports the results of studies concerning an alternative method of obtaining dicarboxylic acids, which consist of the oxidation of cyclic ketones with oxygen or air. The raw materials used were cyclopentanone, cyclo- heptanone, cyclooctanone, cyclododecanone, 1-tetralon, 2-methylhexanone, 3-methylcyclohexanone and 4-methylcy- clohexanone. Oxidation reactions were conducted at 70–100oC, under pressure of 0.1 or 0.4 MPa, for 6 h, utilizing the salts of transition metals as catalyst and acetic acid as solvent. For example, when cyclopentanone was oxidized in the presence of Mn(II) salt, a conversion above 98% and selectivity to glutaric acid up to 68% were obtained. Among synthesized dicarboxylic acids, 1,12-dodecanoic acid was obtained with the highest selectivity of 76%. Keywords: Dicarboxylic acids, cyclic ketones, oxidation, cyclopentanone, glutaric acid. INTRODUCTION dicarboxylic acids, hydrogenation of unsaturated diacids, or esterifi cation of dicarboxylic acids, followed by electrolysis Saturated dicarboxylic acids are an important group and hydrolysis1. The raw materials used in the process of of organic compounds. On an industrial scale, there are forming dicarboxylic acids are primarily obtained from produced acids such as oxalic (600 000 tons/year), malonic benzene (Fig. 1), butadiene (Fig. 2) and higher unsatu- (70 000 tons/year), adipic (2.7 mln tons/year), cork, azelaic rated carboxylic acids occurring naturally (Fig. -

Rules and Regulations Governing Schools in the State of Colorado
COLORADO DEPARTMENT OF PUBLIC HEALTH AND ENVIRONMENT Division of Environmental Health and Sustainability 6 CCR 1010-6 RULES AND REGULATIONS GOVERNING SCHOOLS IN THE STATE OF COLORADO TABLE OF CONTENTS 6.1 Authority .......................................................................................................................................... 2 6.2 Scope and Purpose........................................................................................................................... 2 6.3 Applicability ..................................................................................................................................... 2 6.4 Definitions ........................................................................................................................................ 3 6.5 Incorporation by Reference ............................................................................................................. 7 6.6 Compliance Procedures ................................................................................................................... 7 6.6.1 Inspections ............................................................................................................................. 7 6.6.2 Self-Certification..................................................................................................................... 8 6.6.3 Compliance Assurance ........................................................................................................... 9 6.6.4 Variance Procedures ........................................................................................................... -

The Syntheses and Properties of 1,2-Epoxyalkylphosphonates Bogdan Iorga, Frédéric Eymery, Philippe Savignac
The Syntheses and Properties of 1,2-Epoxyalkylphosphonates Bogdan Iorga, Frédéric Eymery, Philippe Savignac To cite this version: Bogdan Iorga, Frédéric Eymery, Philippe Savignac. The Syntheses and Properties of 1,2- Epoxyalkylphosphonates. SYNTHESIS, Georg Thieme Verlag, 1999, 1999 (02), pp.207-224. 10.1055/s-1999-3378. hal-03161383 HAL Id: hal-03161383 https://hal.archives-ouvertes.fr/hal-03161383 Submitted on 10 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The Syntheses and Properties of 1,2-Epoxyalkylphosphonates Bogdan Iorga, Frédéric Eymery and Philippe Savignac Laboratoire Hétéroéléments et Coordination, UMR CNRS 7653, DCPH, Ecole Polytechnique 91128 Palaiseau Cedex, France.* Abstract. The present review article deals with the methods of preparation of dialkyl 1,2- epoxyalkylphosphonates as well as synthetically useful reactions of this class of compounds. Four main routes to 1,2-epoxyalkylphosphonates are described, in which new developments adapted to each processes are given, including the choice of the leaving group, new cyclisation methods and stereocontrol induction. Thermal and acid catalyzed rearrangements are also covered, and the use of the oxirane ring by nucleophilic opening is highlighted. Particular emphasis is placed on the synthetic approaches to antibiotic fosfomycin.