Reactions of Enolate Ions and Enols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
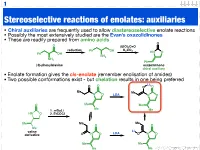
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Alkyl Halides Work Best; Secondary Alkyl Halides Give Lower Yields
CHAPTER 21 ESTER ENOLATES ou have already had considerable experience with carbanionic compounds and their applications in synthetic organic chemistry. The first was acetylide ion in YChapter 9, followed in Chapter 14 by organometallic compounds—Grignard reagents, for example—that act as sources of negatively polarized carbon. In Chapter 18 you learned that enolate ions—reactive intermediates generated from aldehydes and ketones—are nucleophilic, and that this property can be used to advantage as a method for carbon–carbon bond formation. The present chapter extends our study of carbanions to the enolate ions derived from esters. Ester enolates are important reagents in synthetic organic chemistry. The stabilized enolates derived from -keto esters are particularly useful. OO C ␣ C  R CH2 ORЈ -Keto ester: a ketone carbonyl is  to the carbonyl group of the ester. A proton attached to the ␣-carbon atom of a -keto ester is relatively acidic. Typical Ϫ11 acid dissociation constants Ka for -keto esters are Ϸ10 (pKa 11). Because the ␣- carbon atom is flanked by two electron-withdrawing carbonyl groups, a carbanion formed at this site is highly stabilized. The electron delocalization in the anion of a -keto ester is represented by the resonance structures 831 832 CHAPTER TWENTY-ONE Ester Enolates Ϫ Ϫ O O O O O O C C C C C C R C ORЈ R ϪC ORЈ R C ORЈ H H H Principal resonance structures of the anion of a -keto ester We’ll begin by describing the preparation and properties of -keto esters, proceed to a discussion of their synthetic applications, continue to an examination of related species, and conclude by exploring some recent developments in the active field of synthetic car- banion chemistry. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

EI-ICHI NEGISHI Herbert C
MAGICAL POWER OF TRANSITION METALS: PAST, PRESENT, AND FUTURE Nobel Lecture, December 8, 2010 by EI-ICHI NEGISHI Herbert C. Brown Laboratories of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, U.S.A. Not long ago, the primary goal of the synthesis of complex natural products and related compounds of biological and medicinal interest was to be able to synthesize them, preferably before anyone else. While this still remains a very important goal, a number of today’s top-notch synthetic chemists must feel and even think that, given ample resources and time, they are capable of synthesizing virtually all natural products and many analogues thereof. Accepting this notion, what would then be the major goals of organic synthesis in the twenty-first century? One thing appears to be unmistakably certain. Namely, we will always need, perhaps increasingly so with time, the uniquely creative field of synthetic organic and organometallic chemistry to prepare both new and existing organic compounds for the benefit and well-being of mankind. It then seems reasonably clear that, in addition to the question of what compounds to synthesize, that of how best to synthesize them will become increasingly important. As some may have said, the primary goal would then shift from aiming to be the first to synthesize a given compound to seeking its ultimately satisfactory or “last synthesis”. If one carefully goes over various aspects of organic synthetic methodology, one would soon note how primitive and limited it had been until rather recently, or perhaps even today. For the sake of argument, we may propose here that the ultimate goal of organic synthesis is “to be able to synthesize any desired and fundamentally synthesizable organic compounds (a) in high yields, (b) efficiently (in as few steps as possible, for example), (c) selectively, preferably all in t98–99% selectivity, (d) economically, and (e) safely, abbreviated hereafter as the y(es)2 manner.” with or without catalyst R1M + R2X R1R2 + MX R1, R2: carbon groups. -

Enantioselective Solvent-Free Robinson Annulation Reactions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Publications of the IAS Fellows Proc. Indian Acad. Sci. (Chem. Sci.), Vol. 113, No. 3, June 2001, pp 197–213 Ó Indian Academy of Sciences Enantioselective solvent-free Robinson annulation reactions D RAJAGOPAL, R NARAYANAN and S SWAMINATHAN* Department of Organic Chemistry, University of Madras, Guindy Campus, Chennai 600 025, India e-mail: [email protected] MS received 28 March 2001 Abstract. The enantioselective cyclization of the prochiral cyclic substrates 1 to 7 and 26, can be carried out in the neat using S-proline as catalyst. The substrates 18 to 22 and 27 could not be cyclized with S-proline but could be cyclized with a mixture of S-phenylalanine and d-camphorsulphonic acid. The enantioselective cyclization of prochiral acyclic triones 45 and 47 and also the racemic tricarbonyl compounds 54 to 57 could also be carried out in the neat using S-proline as catalyst. The optically active enediones obtained in the above cyclizations could also be obtained directly from 1,3-diones or 2-hydroxymethylene cycloalkanones in a one-pot reaction with methyl vinyl ketone (MVK) and S-proline in the absence of solvents. 13C NMR studies of the one-pot synthesis of S-11 and S-14 reveal that the annulations involve initial formation of an acid-base complex followed by a Michael reaction and then an enantioselective cyclization. Such enantioselective cyclizations probably occur on the surface of S-proline crystals. Keywords. Enantioselective annulation; cyclization; S-proline; S-phenylalanine; d-camphorsulphonic acid. -

Robinson Annulation
Robinson annulation The Robinson annulation is a chemical reaction used in organic Robinson annulation chemistry for ring formation. It was discovered by Robert Robinson Named after Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds.[1] The method uses a ketone and a methyl Reaction type Ring forming vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring reaction by a Michael addition followed by an aldol condensation. This Identifiers procedure is one of the key methods to form fused ring systems. Organic robinson-annulation Chemistry Portal RSC ontology RXNO:0000380 ID Formation of cyclohexenone and derivatives are important in chemistry for their application to the synthesis of many natural products and other interesting organic compounds such as antibiotics and steroids.[2] Specifically, the synthesis of cortisone is completed through the use of the Robinson annulation.[3] The initial paper on the Robinson annulation was published by William Rapson and Robert Robinson while Rapson studied at Oxford with professor Robinson. Before their work, cyclohexenone syntheses were not derived from the α,β-unsaturated ketone component. Initial approaches coupled the methyl vinyl ketone with a naphthol to give a naphtholoxide, but this procedure was not sufficient to form the desired cyclohexenone. This was attributed to unsuitable conditions of the reaction.[1] Robinson and Rapson found in 1935 that the interaction between cyclohexanone and α,β-unsaturated ketone afforded the -

Diversity-Oriented Combinatorial Biosynthesis of Benzenediol Lactone Scaffolds by Subunit Shuffling of Fungal Polyketide Synthases
Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases Yuquan Xua,b,1, Tong Zhouc,1, Shuwei Zhangc, Patricia Espinosa-Artilesb, Luoyi Wangc, Wei Zhanga, Min Lina, A. A. Leslie Gunatilakab,d, Jixun Zhanc,2, and István Molnárb,d,2 aBiotechnology Research Institute, The Chinese Academy of Agricultural Sciences, Beijing 100081, P. R. China; bNatural Products Center, School of Natural Resources and the Environment, University of Arizona, Tucson, AZ 85706; cDepartment of Biological Engineering, Utah State University, Logan, UT 84322; and dBio5 Institute, University of Arizona, Tucson, AZ 85721 Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved June 23, 2014 (received for review April 16, 2014) Combinatorial biosynthesis aspires to exploit the promiscuity of biosynthesis inhibitory activities in animals. 10,11-dehydrocurvularin microbial anabolic pathways to engineer the synthesis of new (7;Fig.1)isaDALwitha12-memberedring(DAL12)that chemical entities. Fungal benzenediol lactone (BDL) polyketides modulates the heat shock response and the immune system (8, 9). are important pharmacophores with wide-ranging bioactivities, BDL scaffolds are biosynthesized by pairs of collaborating, including heat shock response and immune system modulatory sequentially acting iterative polyketide synthases (iPKSs) (3) – effects. Their biosynthesis on a pair of sequentially acting iterative forming quasi-modular BDL synthases (BDLSs) (Fig. 1) (11 14). polyketide synthases (iPKSs) offers a test case for the modulariza- Each of the BDLS subunits catalyze recursive, decarboxylative tion of secondary metabolic pathways into “build–couple–pair” Claisen condensations of malonyl-CoA using a single core set of ketoacyl synthase (KS), acyl transferase (AT), and acyl carrier combinatorial synthetic schemes. -

Experiment 19 — Aldol Condensation
Chem 22 Spring 2010 Experiment 19 — Aldol Condensation _____________________________________________________________________________ Pre-lab preparation. (1) Write the mechanism of the base-catalyzed aldol condensation of acetone and a generalized aromatic aldehyde, Ar–CH=O to give the α,β-unsaturated product (i.e. the reaction at the top of the next page, but with a 1:1 ratio of ketone and aldehyde). Remember that dehydration in this case occurs under basic conditions, so it can't start with protonation of the hydroxyl group. Nor can it go via an E2 pathway. (2) Draw the structures of all the possible aldehyde and ketone reactants (not the 25 possible condensation products!). (3) The new CC double bonds of the condensation products are E rather than Z, as shown in the acetone example. Why? (4) What's the purpose of rinsing the crude product with dilute acetic acid, followed by ethanol? (5) What is the procedure for carrying out a single-solvent recrystallization? Write this out in detail. The aldol condensation has historically been one of the favorite tools in the synthetic organic chemist's repertoire because of its versatility in forming new CC bonds. Since its discovery in the 1870s the aldol condensation has been use extensively in the synthesis of natural products and other complex molecules. In a typical base-catalyzed aldol condensation an enolate ion attacks the carbonyl group of an aldehyde or ketone. This carbonyl addition produces a β-hydroxy carbonyl compound. In many cases the initially formed condensation product undergoes an "E1cB" dehydration to produce an α,β-unsaturated carbonyl compound as the final product. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds
http://dx.doi.org/10.5935/0103-5053.20140255 J. Braz. Chem. Soc., Vol. 25, No. 12, 2186-2214, 2014. Printed in Brazil - ©2014 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00 Review Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds André F. P. Biajoli, Cristiane S. Schwalm, Jones Limberger, Thiago S. Claudino and Adriano L. Monteiro* Laboratory of Molecular Catalysis, Institute of Chemistry, UFRGS, Av. Bento Gonçalves, 9500, 91501-970 Porto Alegre-RS, Brazil A grande capacidade do paládio de formar ligações carbono-carbono entre substratos apropriadamente funcionalizados permitiu que os químicos orgânicos efetuassem transformações antes impossíveis ou alcançáveis somente através de rotas muito longas. Neste contexto, uma das mais elegantes e importantes aplicações das reações de acoplamento cruzado catalisadas por paládio é a síntese de compostos de interesse farmacêutico. A presente revisão tem por objetivo apresentar uma visão geral do uso de acoplamentos cruzados na síntese de componentes de medicamentos (ou de candidatos a medicamentos), independentemente da escala, compreendendo o período de 2011 até o final de julho de 2014. The impressive ability of palladium to assemble C-C bonds between appropriately functionalized substrates has allowed synthetic organic chemists to perform transformations that were previously impossible or only possible using multi-step approaches. In this context, one of the most important and elegant applications of the Pd-catalyzed C-C coupling reactions currently is the synthesis of pharmaceuticals. This review is intended to give a picture of the applications of Pd-catalyzed C-C cross-coupling reactions for the synthesis of drug components or drug candidates regardless of the scale from 2011 through to the end of July, 2014. -

The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis
GENERAL ARTICLE The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis Gopalpur Nagendrappa and Y C Sunil Kumar The 2010 Nobel Prize in Chemistry was awarded to three scientists, R F Heck, E-I Negishi and A Suzuki, for their work on “Palladium – Catalyzed Cross Couplings in Organic Syn- (left) G Nagendrappa thesis”. It pertains to research done over a period of four was a Professor of decades. The synthetic procedures embodied in their work Organic Chemistry at enable construction of C–C bond selectively between complex Bangalore University, molecules as in simple ones at desired positions without and Head of the Department of Medici- disturbing any functional groups at other parts of the reacting nal Chemistry, Sri molecules. The work finds wide applications in the synthesis Ramachandra (Medical) of pharmaceuticals, agricultural chemicals, and molecules for University, Chennai. electronics and other applications. It would not have been He is currently in Jain University, Bangalore. possible to synthesize some of the complex natural products or He continues to teach synthetic compounds without using these coupling reactions and do research. His in one or more steps. work is in the area of organosilicon chemis- Introduction try, synthetic and mechanistic organic In mythical stories and folk tales we come across characters that, chemistry, and clay- catalysed organic while uttering some manthras (words of charm), throw a pinch or reactions (Green fistful of a magic powder, and suddenly there appears the object Chemistry). or person they wished for or an event happens the way they want. (right) Sunil Kumar is A large number of movies have been made with such themes and a PhD from Mysore characters that would be depicted as ‘scientists’. -

Aldol Condensation- Aldehyde (Or Ketone) Enolate Condenses with Aldehyde (Or Ketone)
Chem 232 Summary of Alpha Substitutions page 1 Aldol Condensation- aldehyde (or ketone) enolate condenses with aldehyde (or ketone): O CH O O H 3 H CH 3 CH3 C C C C CH C C CH2 CH2 H -H O H H O OH 2 H nucleophile electrophile -hydroxy aldehyde -unsaturated aldehyde The nucleophile can be a ketone enolate or aldehyde enolate and the electrophile (shaded) can be an aldehyde or ketone. Crossed Aldol- Condensation between two different carbonyls. The component without hydrogens is the electrophile: O O O OH O C CH C C CH CH3 C H CH -H2O CH 2 CH3 H 3 ketone enolate no -hydrogens -unsaturated ketone Aldol Cyclizations- A dicarbonyl produces an enolate and carbonyl in the same molecule: enolate from a O OH 1,5-diketone CH OH 3 CH3 O O -H2O O CH2 CH3 CH3 CH2 O Claisen Condensation- Similar to Aldol condensation except the nucleophile is an ester enolate; O O O O O O + EtO C CH C OEt EtO C CH2 C EtO C CH2 CH3 C OEt 2 ketoester CH3 CH3 Dieckmann Cyclization- Internal Claisen condensation similar to Aldol cyclization. A 1,6 diester gives a 5-membered ring and a 1,7 diester gives a 6-membered ring: O OEt O OEt cyclic ketoester C C OEt O O Crossed Claisen- Similar to crossed Aldol- Electrophile has nohydrogens: O O O O C C EtO EtO C CH2 EtO C CH2 ketoester Variations on Crossed Claisen- ketone enolate and ester condensation. Esters, carbonates, formates and oxalates are common electrophiles: O O O O O O O O H C OEt H EtO C OEt OEt ethyl formate -ketoaldehyde diethyl carbonate -ketoester O O O O O OEt diketoester EtO C C OEt O diethyl oxalate