Aldol Reactions: E-Enolates and Anti-Selectivity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download (1MB)
THE IN VITRO METABOLISM OF THREE ANTICANCER DRUGS by Yun Fan B.S., West China University of Medical Sciences, 1996 M.S., West China University of Medical Sciences, 2001 Submitted to the Graduate Faculty of School of Pharmacy in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2007 UNIVERSITY OF PITTSBURGH SCHOOL OF PHARMACY This dissertation was presented by Yun Fan It was defended on March 23, 2007 and approved by Samuel M. Poloyac, Assistant Professor, Pharmaceutical Sciences Raman Venkataramanan, Professor, Pharmaceutical Sciences Jack C. Yalowich, Associate Professor, Pharmacology Michael A. Zemaitis, Professor, Pharmaceutical Sciences Billy W. Day, Professor, Pharmaceutical Sciences ii Copyright © by Yun Fan 2007 iii THE IN VITRO METABOLISM OF THREE ANTICANCER DRUGS Yun Fan, Ph.D. University of Pittsburgh, 2007 Etoposide is a widely used topoisomerase II inhibitor particularly useful in the clinic for treatment of disseminated tumors, including childhood leukemia. However, its use is associated with the increased risk of development of secondary acute myelogenous leukemias. The mechanism behind this is still unclear. It was hypothesized that etoposide ortho-quinone, a reactive metabolite previously shown to be generated in vitro by myeloperoxidase, the major oxidative enzyme in the bone marrow cells from which the secondary leukemias arise, might be a contributor to the development of treatment-related secondary leukemias. Experiments showed that the glutathione adduct of etoposide ortho-quinone was formed in myeloperoxidase- expressing human myeloid leukemia HL60 cells treated with etoposide, that its formation was enhanced by addition of the myeloperoxidase substrate hydrogen peroxide, and that the glutathione adduct level was dependent on myeloperoxidase. -

Optimizing the Least Nucleophilic Anion. a New, Strong Methyl+ Reagent Daniel Stasko and Christopher A
Published on Web 01/25/2002 Optimizing the Least Nucleophilic Anion. A New, Strong Methyl+ Reagent Daniel Stasko and Christopher A. Reed* Department of Chemistry, UniVersity of California, RiVerside, California 92521-0304 Received August 3, 2001 The ideal weakly coordinating counterion for reactive cations would be the least nucleophilic, least basic, most inert anion available. It should also be inexpensive, have useful spectroscopic handles, and confer good solubility and crystallizing properties on - its salts. Triflate anion, CF3SO3 , has served chemistry well in this regard but larger size, absence of lone pairs, and extreme chemical inertness have recently made carboranes1 or fluorinated tetraphe- nylborates the preferred choice for many applications.2 These anions have led to the solution of the silylium ion problem,3 enhanced Lewis acid catalysis by Li+ ion,4-6 new “strong-but-gentle” superacids,7 commercially viable olefin polymerization catalysts,8 new possibilities for electrolytes9,10 and ionic liquids,11 and the •+ 7 + 12 isolation of new reactive cations such as C60 , Bu3Sn , Cu- + 13 (CO)4 , etc. Figure 1. The 1-H-2,3,4,5,6-pentamethyl-7,8,9,10,11,12-hexahalo-CB11 - ) The perfluorinated tetraphenylborate anion is the preferred choice carborane anions, 1-H-CB11Me5X6 (X Cl, Br, I), used in this work. for price and solubility reasons but its salts frequently form liquid 29 Table 1. Selected Si Chemical Shifts (ppm) for i-Pr3SiY clathrates and fail to crystallize. The boron-phenyl bond is rather 29 easily cleaved by strong Brønsted14 or Lewis15 acids. Halogenated compd Si conditions carborane anions are much more inert than tetraphenylborates, their i-Pr3Si(1-H-CB11H5Br6) 100 C6D6 salts crystallize well, but they are more expensive and their salts i-Pr3Si(1-H-CB11H5Cl6) 103 C6D6 i-Pr Si(F -BPh )a 108 solid state less soluble. -
Stereoselective Reactions of Enolates: Auxiliaries
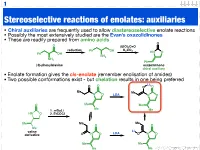
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Nicole Goodwin Macmillan Group Meeting April 21, 2004
Discodermolide A Synthetic Challenge Me Me Me HO Me O O OH O NH2 Me Me OH O Me Me OH Nicole Goodwin MacMillan Group Meeting April 21, 2004 Me Me Me HO Discodermolide Me O O OH O NH2 Isolation and Biological Activity Me Me OH O Me Me OH ! Isolated in 1990 by the Harbor Branch Oceanographic Institution from the Caribbean deep-sea sponge Discodermia dissoluta ! Not practical to produce discodermolide from biological sources ! 0.002% (by mass) isolation from frozen sponge ! must be deep-sea harvested at a depth in excess of 33 m ! Causes cell-cycle arrest at the G2/M phase boundary and cell death by apoptosis ! Member of an elite group of natural products that act as microtubule-stabilizing agent and mitotic spindle poisons ! Taxol, epothilones A and B, sarcodictyin A, eleutherobin, laulimalide, FR182877, peloruside A, dictyostatin ! Effective in Taxol-resistant carcinoma cells ! presence of a small concentration of Taxol amplified discodermolide's toxicity by 20-fold ! potential synergies with the combination of discodermolide with Taxol and other anticancer drugs ! Licensed by Novartis from HBOI in 1998 as a new-generation anticancer drug 1 Cell Apoptosis M - Mitosis G0 Discodermolide disrupts the G2/M phases of the cell cycle. Resting Discodermolide, like Taxol, inhibits microtubule depolymerization by binding G1 - Gap 1 to tubulin. G2 - Gap 2 cells increase in size, cell growth control checkpoint no microtubules = no spindle fiber new proteins formation S - Synthesis DNA replication Me Me Me HO Discodermolide Me O O OH O NH2 Isolation -

United States Patent (19) 11 Patent Number: 5,789,605 Smith, I Et Al
IIIUSOO5789605A United States Patent (19) 11 Patent Number: 5,789,605 Smith, I et al. 45) Date of Patent: Aug. 4, 1998 54 SYNTHETICTECHNIQUES AND Jacquesy et al., "Metabromation Du Dimethyl-2, 6 Phenol INTERMEDIATES FOR POLYHYDROXY, Et De Son Ether Methylique En Milieu Superacide", Tetra DENYL LACTONES AND MIMCS hedron, 1981, 37, 747-751. THEREOF Kim et al., "Conversion of Acetals into Monothioacetals, O-Alkoxyazides and O-Alkoxyalkyl Thioacetates with 75) Inventors: Amos B. Smith, III. Merion; Yuping Magnesium Bromide". Tetra, lett, 1989, 30(48), Qiu; Michael Kaufman, both of 6697-6700. Philadelphia; Hirokaza Arimoto, Longley et al., "Discodermolide-A New, Marine-Derived Drexel Hill, all of Pa.; David R. Jones, Immunosuppressive Compound". Transplantation, 1991. Milford, Ohio; Kaoru Kobayashi, 52(4) 650-656. Osaka, Japan Longley et al., "Discodermolide-A New, Marine-Derived Immunosuppressive Compound". Transplantation, 1991. 73 Assignee: Trustees of the University of 52(4), 657-661. Pennsylvania, Philadelphia, Pa. Longley et al., "Immunosuppression by Discodermolide". Ann. N.Y. Acad. Sci., 1993. 696, 94-107. (21) Appl. No.:759,817 Nerenburg et al., "Total Synthesis of the Immunosuppres 22 Filed: Dec. 3, 1996 sive Agent (-)-Discodermolide". J. Am. Chem, Soc., 1993, 115, 12621-12622. (51) Int. Cl. ................. C07D407/06; C07D 319/06; Paterson, I. et al., "Studies Towards the Total Synthesis of CO7D 309/10 the Marine-derived Immunosuppressant Discodermolide; (52) U.S. Cl. .......................... 549/370; 549/374; 549/417 Asymmetric Synthesis of a C-Cö-Lactone Subunit". J. (58) Field of Search ..................................... 549/370,374, Chen. Soc. Chen, Commun., 1993, 1790-1792. 549/417 Paterson, I. et al., "Studies Towards the Total Synthesis of the Marine-derived Immunosuppresant Discodermolide; 56 References Cited Asymmetric Synthesis of a C-C Subunit". -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Dissertation.Pdf (1.198Mb)
Syntheses and Bioactivities of Targeted and Conformationally Restrained Paclitaxel and Discodermolide Analogs Chao Yang Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University In the partial fulfillment of the requirement for the degree of Doctor of Philosophy In Chemistry Dr. David G. I. Kingston, Chairman Dr. Karen Brewer Dr. Paul R. Carlier Dr. Felicia. Etzkorn Dr. Harry W. Gibson August 26 2008 Blacksburg, Virginia Keywords: Taxol, drug targeting, , thio-taxol, tubulin-binding conformation, , T-taxol conformation, macrocyclic taxoids, discodermolide Copyright 2008, Chao Yang Syntheses and Bioactivities of Targeted and Conformationally Restrained Paclitaxel and Discodermolide Analogs Chao Yang Abstract Paclitaxel was isolated from the bark of Taxus brevifolia in the late 1960s. It exerts its biological effect by promoting tubulin polymerization and stabilizing the resulting microtubules. Paclitaxel has become one of the most important current drugs for the treatment of breast and ovarian cancers. Studies aimed at understanding the biologically active conformation of paclitaxel bound on β–tubulin are described. In this work, the synthesis of isotopically labeled taxol analogs is described and the REDOR studies of this compound complexed to tubulin agrees with the hypothesis that palictaxel adopts T-taxol conformation. Based on T-taxol conformation, macrocyclic analogs of taxol have been prepared and their biological activities were evaluated. The results show a direct evidence to support T-taxol conformation. (+) Discodermolide is a polyketide isolated from the Caribbean deep sea sponge Discodermia dissoluta in 1990. Similar to paclitaxel, discodermolide interacts with tubulin and stabilizes the microtubule in vivo. Studies aimed at understanding the biologically active conformation of discodermolide bound on β–tubulin are described. -

Total Synthesis of the Marine Natural Product (+)-Discodermolide in Multigram Quantities*
Pure Appl. Chem., Vol. 79, No. 4, pp. 685–700, 2007. doi:10.1351/pac200779040685 © 2007 IUPAC Total synthesis of the marine natural product (+)-discodermolide in multigram quantities* Stuart J. Mickel Novartis Pharma AG, CHAD, WKL-684.2.31 Klybeckstrasse, 4052 Basel, Switzerland Abstract: The novel polyketide (+)-discodermolide was isolated in very small quantities from sponge extracts. This compound is one of several microtubule stabilizers showing promise as novel chemotherapeutic agents for the treatment of cancer. The clinical evaluation of this and similar compounds is hampered by lack of material, and at present, the only way to obtain the necessary quantities is total chemical synthesis. Keywords: discodermolide; boron aldol reaction; isolation; side products; olefination. INTRODUCTION The marine natural product (+)-discodermolide 1 was isolated by workers at the Harbor Branch Oceanographic Institute from the deep-sea sponge Discodermia dissoluta in 1990 [1]. It is obtained from the sponge in extremely small quantities, 0.002 % in the frozen material. This compound is one of several natural products exhibiting inhibition of microtubule depolymerization and as such shows considerable promise as a novel chemotherapeutic agent for the treatment of cancer [2]. In contrast to Taxol®, another microtubule stabilizer, (+)-1 shows activity in the Taxol-resistant cell lines which over- express P-glycoprotein, the multidrug-resistant transporter. The similarities and differences between (+)-1 and Taxol have recently been discussed in detail [3]. The structure of (+)-1 consists of a linear polypropionate chain interrupted by 3 cis-olefins. It con- tains 13 chiral centers, 6 of which are hydroxyl groups, one esterified as a lactone, another as a car- bamic acid ester. -

Marine Natural Products: a Source of Novel Anticancer Drugs
marine drugs Review Marine Natural Products: A Source of Novel Anticancer Drugs Shaden A. M. Khalifa 1,2, Nizar Elias 3, Mohamed A. Farag 4,5, Lei Chen 6, Aamer Saeed 7 , Mohamed-Elamir F. Hegazy 8,9, Moustafa S. Moustafa 10, Aida Abd El-Wahed 10, Saleh M. Al-Mousawi 10, Syed G. Musharraf 11, Fang-Rong Chang 12 , Arihiro Iwasaki 13 , Kiyotake Suenaga 13 , Muaaz Alajlani 14,15, Ulf Göransson 15 and Hesham R. El-Seedi 15,16,17,18,* 1 Clinical Research Centre, Karolinska University Hospital, Novum, 14157 Huddinge, Stockholm, Sweden 2 Department of Molecular Biosciences, the Wenner-Gren Institute, Stockholm University, SE 106 91 Stockholm, Sweden 3 Department of Laboratory Medicine, Faculty of Medicine, University of Kalamoon, P.O. Box 222 Dayr Atiyah, Syria 4 Pharmacognosy Department, College of Pharmacy, Cairo University, Kasr el Aini St., P.B. 11562 Cairo, Egypt 5 Department of Chemistry, School of Sciences & Engineering, The American University in Cairo, 11835 New Cairo, Egypt 6 College of Food Science, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China 7 Department of Chemitry, Quaid-i-Azam University, Islamabad 45320, Pakistan 8 Department of Pharmaceutical Biology, Institute of Pharmacy and Biochemistry, Johannes Gutenberg University, Staudingerweg 5, 55128 Mainz, Germany 9 Chemistry of Medicinal Plants Department, National Research Centre, 33 El-Bohouth St., Dokki, 12622 Giza, Egypt 10 Department of Chemistry, Faculty of Science, University of Kuwait, 13060 Safat, Kuwait 11 H.E.J. Research Institute of Chemistry, -

Predicting Glycosylation Stereoselectivity Using Machine Learning† Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Predicting glycosylation stereoselectivity using machine learning† Cite this: Chem. Sci.,2021,12,2931 ab a ab All publication charges for this article Sooyeon Moon, ‡ Sourav Chatterjee, ‡ Peter H. Seeberger have been paid for by the Royal Society and Kerry Gilmore §*a of Chemistry Predicting the stereochemical outcome of chemical reactions is challenging in mechanistically ambiguous transformations. The stereoselectivity of glycosylation reactions is influenced by at least eleven factors across four chemical participants and temperature. A random forest algorithm was trained using a highly reproducible, concise dataset to accurately predict the stereoselective outcome of glycosylations. The steric and electronic contributions of all chemical reagents and solvents were quantified by quantum mechanical calculations. The trained model accurately predicts stereoselectivities for unseen nucleophiles, electrophiles, acid catalyst, and solvents across a wide temperature range (overall root mean square error 6.8%). All predictions were validated experimentally on a standardized microreactor platform. The model helped to identify novel ways to control glycosylation stereoselectivity and Creative Commons Attribution 3.0 Unported Licence. Received 11th November 2020 accurately predicts previously unknown means of stereocontrol. By quantifying the degree of influence Accepted 24th December 2020 of each variable, we begin to gain a better general understanding of the transformation, for example that DOI: 10.1039/d0sc06222g environmental factors influence the stereoselectivity of glycosylations more than the coupling partners in rsc.li/chemical-science this area of chemical space. Introduction retrosynthesis,9 reaction performance10 and products,11,12 the identication of new materials and catalysts,13–15 as well as Predicting the outcome of an organic reaction generally enantioselectivity16,17 have been addressed. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Stereoselectivity in Atmospheric Autoxidation Kristian H
Subscriber access provided by Caltech Library Clusters, Radicals, and Ions; Environmental Chemistry Stereoselectivity in Atmospheric Autoxidation Kristian H. Møller, Eric Joseph Praske, Lu Xu, John D. Crounse, Paul O. Wennberg, and Henrik Grum Kjaergaard J. Phys. Chem. Lett., Just Accepted Manuscript • DOI: 10.1021/acs.jpclett.9b01972 • Publication Date (Web): 23 Sep 2019 Downloaded from pubs.acs.org on September 24, 2019 Just Accepted “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.