Page 1 of 108 RSC Advances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Stereoselective Reactions of Enolates: Auxiliaries
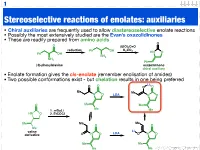
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -
Chiral Auxiliaries and Optical Resolving Agents
Chiral Auxiliaries and Optical Resolving Agents Most bioactive substances are optically active. For instance, if This brochure introduces a variety of chiral auxiliaries and a substance is synthesized as a racemic compound, its optical resolving agents. We hope that it will be useful for your enantiomer may show no activity or even undesired bioactivity. research of the synthesis of optically active compounds. Thus, methods to gain enantiopure compounds have been Additionally, TCI has some brochures introducing chiral developed. When synthesizing enantiopure compounds, the compounds for the chiral pool method in “Chiral Building Blocks”, methods are roughly divided into three methods. “Terpenes”, “Amino Acids” and other brochures. Sugar derivatives are also introduced in a catalog, “Reagents for Glyco Chemistry Chiral pool method: & Biology”, and category pages of sugar chains. Furthermore, The method using an easily available chiral compound as a TCI has many kinds of catalysts for asymmetric synthesis and starting material like an amino acid or sugar. introduce them in brochures such as “Asymmetric Synthesis” and Asymmetric synthesis: “Asymmetric Organocatalysts”, and other contents. The method to introduce an asymmetric point to compounds You can search our information through “asymmetric synthesis” without an asymmetric point. Syntheses using achiral as a keyword. auxiliaries are included here. Optical resolution: The method to separate a racemic compound into two ● Reactions with Chiral Auxiliaries enantiomers. The direct method using a chiral column and One of the most famous named reactions using chiral auxiliaries1) the indirect method to separate two enantiomers using is the Evans aldol reaction.2) This reaction is quite useful because optical resolving agents to convert into diastereomers are this reaction can efficiently introduce two asymmetric carbons into examples. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

1,2-Asymmetric Induction in Carbonyl Compounds - a Computational Study
1,2-Asymmetric Induction in Carbonyl Compounds - A Computational Study by Jeffrey Retallick B.Sc., University of New Brunswick, 2015 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science In the Graduate Academic Unit of Chemistry Supervisor(s): Ghislain Deslongchamps, Ph.D., Chemistry Examining Board: Rodney Cooper, M.Sc., Computer Science, Chair External Examiner: Ben Newling, Ph.D., Physics This thesis is accepted by the Dean of Graduate Studies THE UNIVERSITY OF NEW BRUNSWICK October, 2019 © Jeffrey Retallick, 2020 Abstract In asymmetric synthesis, it is important to reliably predict the major stereoisomeric prod- uct of a reaction. One such reaction is the nucleophilic addition to a carbonyl compound featuring an adjacent chiral carbon. Several reaction models exist in literature to predict the facial selectivity of these reactions. These models provide simple visual drawings to quickly predict the major product of such a reaction without requiring exhaustive quantum mechanical calculations. These models are used on a daily basis, and some models per- form better than others, making it valuable to investigate which ones are the most effective. For the first time in this thesis, high-level computations have been performed on all of the literature models to verify their efficacy. The results of this thesis offers a definitive answer that the Felkin-Anh and Wintner models are the most effective, and that bent bond theory offers an interesting insight on the mechanics of these reactions. ii Dedication Kyle, Nan, and Taryn. iii Acknowledgements Thanks to my family for their support. Ghislain, thank you for putting up with me over the past while. -

Chapter 8. Chiral Catalysts José M
Chapter 8. Chiral Catalysts José M. Fraile, José I. García, José A. Mayoral 1. The Origin of Enantioselectivity in Catalytic Processes: the Nanoscale of Enantioselective Catalysis. Enantiomerically pure compounds are extremely important in fields such as medicine and pharmacy, nutrition, or materials with optical properties. Among the different methods to obtain enantiomerically pure compounds, asymmetric catalysis1 is probably the most interesting and challenging, in fact one single molecule of chiral catalyst can transfer its chiral information to thousands or even millions of new chiral molecules. Enantioselective reactions are the result of the competition between different possible diastereomeric reaction pathways, through diastereomeric transition states, when the prochiral substrate complexed to the chiral catalyst reacts with the corresponding reagent. The efficiency of the chirality transfer, measured as enantiomeric excess [% ee = (R−S)/(R+S) × 100], depends on electronic and steric factors in a very subtle form. A simple calculation shows that differences in energy of only 2 kcal/mol between these transition states are enough to obtain more than 90% ee, and small changes in any of the participants in the catalytic process can modify significantly this difference in energy. Those modifications may occur in the near environment of the catalytic centre, at less than 1 nm scale, but also at longer distances in the catalyst, substrate, reagent, solvent, or support in the case of immobilized catalysts. This is the reason because asymmetric -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

APPLICATIONS in ASYMMETRIC SYNTHESIS Carlos
324 SYNTHESIS OF OCTAHYDROBENZO - 1, 2,3 - DIAZAPHOSPHOLIDINE - 2 - OXIDES AND THEIR DERIVATIVES: APPLICATIONS IN ASYMMETRIC SYNTHESIS DOI: http://dx.medra.org/ 10.17374/targets.2020.23.324 Carlos Cruz - Hernández a , José M. Landeros a , Eusebio Juaristi * a,b a Departamento de Química, Centro de Investigación y de E studios Avanzados, Avenida IPN 2508, 07360 Ciudad de México, Mexico b El Colegio Nacional, Luis González Obregón 23, Centro Histórico, 06020 Ciudad de México, Mexico (e - mail: [email protected]; [email protected]) Abstract. This chapter outlines recent efforts devoted to the synthesis of heterocycles that include the octahydrobenzo - 1,3,2 - diazaphospholidine - 2 - oxide fragment, as well as their application in asymmetri c synthesis. The first part of this review provides a brief discussion of the general structural characteristics of this phosphorus - containing heterocyclic scaffold. The second part describes the synthetic paths that were undertaken to synthesize the desir ed heterocycles , as well as some relevant considerations pertaining the spectroscopic characterization of the phosphorus - containing heterocycles of interest . The third part provides several illustrative examples where the novel chiral heterocycles were employed in ena ntioselective synthesis. The new phosphorus - containing heterocycles proved useful : i) as chiral auxiliaries in nucleophilic addition reactions, as well as as imine activators in electrophilic addition reactions; ii ) as c hiral ligands i n nucleophilic allylation and crotonylation of prochiral aldehydes , and iii) as c hiral organocatalysts in enantioselective aldol, Michael, and cascade reactions. Contents 1. Introduction 2. Synthesis of the octahydrobenzo - 1,3,2 - diazaphospho lidine - 2 - oxide s 2.1 . Conformational and configurational assignments 3 . -

Application of Chiral Sulfoxides in Asymmetric Synthesis
MOJ Bioorganic & Organic Chemistry Review Article Open Access Application of chiral sulfoxides in asymmetric synthesis Abstract Volume 2 Issue 2 - 2018 Chiral sulfoxides are used as a toolbox for the synthesis of enantiomeric/diastereomeric compounds, which are used as precursors for the pharmaceutically/chemically Ganapathy Subramanian Sankaran,1 important molecules. The current review focuses on applying these chiral sulfoxides Srinivasan Arumugan,2 Sivaraman towards the synthesis of the compounds having stereogenic center. In general, the 3 stereogenic center induced by the sulfoxide is able to direct the stereochemistry of Balasubramaniam 1University of Massachusetts Medical School, USA further transformation necessary to complete the total synthesis of bioactive molecules. 2Department of Science and Humanity (Chemistry), Karunya The nature of the reactive conformation of the sulfoxide is strongly dependent on the Institute of Technology and Sciences, India nature of the substituents at C-α and/or C-β. 3Indian Institute of Technology Madras, Chennai, India Correspondence: Sivaraman Balasubramaniam, Senior Research Scientist, Indian Institute of Technology Madras, Chennai, India, Tel +9177 1880 5113, Email [email protected] Received: March 07, 2018 | Published: March 29, 2018 Introduction occurred in a further oxidation step of one of the sulfinyl enantiomer to sulfone.13 The titanium-binaphthol complex catalyzes not only the Over the last three decades, the sulfinyl group has received asymmetric oxidation but also the kinetic -

Kem-4.450 Asymmetric Synthesis
KemKem--4.4504.450 AsymmetricAsymmetric SynthesisSynthesis Prof. Ari Koskinen Laboratory of Organic Chemistry © Helsinki University of Technology, Laboratory of Organic Chemistry © Ari Koskinen, 2007 ChiralityChirality andand DifferingDiffering PropertiesProperties OO Carvone spearmint odor caraway NH NH 2 H 2 H Aspartame HO2C NCO2Me HO2C NCO2Me (Nutrasweet) O O sweet bitter O O Thalidomide N N O O OON OON H H sedative, hypnotic teratogenic © Helsinki University of Technology, Laboratory of Organic Chemistry © Ari Koskinen, 2007 PharmaceuticalsPharmaceuticals Growing need for enantiopure compounds Enantiomers/diastereomers may have adverse effects Diastereomers usually easier to separate Enantiomers: FDA required © Helsinki University of Technology, Laboratory of Organic Chemistry © Ari Koskinen, 2007 PharmaceuticalsPharmaceuticals Penicillamine: NH2 NH2 CO2H CO2H SH SH antidote for Pb, Au, Hg can cause optic atrophy => blindness Timolol: O OH O OH H H N O N N O N N N N N S S adrenergic blocker ineffective © Helsinki University of Technology, Laboratory of Organic Chemistry © Ari Koskinen, 2007 SalesSales ofof EnantiomericEnantiomeric DrugsDrugs andand IntermediatesIntermediates Chem. Eng. News 2001, 79 (40), 79-97. © Helsinki University of Technology, Laboratory of Organic Chemistry © Ari Koskinen, 2007 AsymmetricAsymmetric InductionInduction -- DefinitionsDefinitions Chirality - handedness Asymmetry - lacking all symmetry (except E) B Dissymmetry - lacking some element of symmery H NB!!! Molecules can be chiral but not asymmetric! -

Catalytic Asymmetric Synthesis
Catalytic Asymmetric Synthesis Dr Alan Armstrong Rm 313 (RCS1), ext. 45876; [email protected] 7 lectures Recommended texts: “Catalytic Asymmetric Synthesis, 2nd edition”, ed. I Ojima, Wiley-VCH, 2000 (or 1st ed., 1993, which contains most of the lecture material) “Asymmetric Synthesis”, G Procter, Oxford, 1996 Aims of course: To give students an understanding of the basic principles of asymmetric catalysis, and to demonstrate these in the context of state-of-the-art catalytic asymmetric processes for C-C bond formation and redox processes. Course objectives: At the end of this course you should be able to: • Recognise the types of functional groups which can be prepared by catalytic asymmetric methods discussed in the course; • Use this knowledge in planning the synthesis of enantiomerically enriched compounds from given prochiral starting materials; • Outline the scope and limitations of any methods you propose, with respect to parameters such as turnover, substrate and functional group tolerance, availability of catalysts and/or ligands etc Course content (by lecture, approximately): 1. Introduction and general principles of asymmetric catalysis. Oxidation of functionalised olefins: asymmetric epoxidation of allylic alcohols and enones 2. Oxidation of unfunctionalised olefins: epoxidation, dihydroxylation and aminohydroxylation. 3. Reduction of olefins: asymmetric hydrogenation. 4. Reduction of ketones and imines 5. Asymmetric C-C bond formation: nucleophilic attack on carbonyls, imines and enones 6. Asymmetric transition metal catalysis in C-C bond forming reactions (cross-couplings, Heck reactions, allylic substitution, carbenoid reactions) 7. Chiral Lewis acid catalysis (aldol reactions, cycloadditions, Mannich reactions, allylations) 1 Catalytic Asymmetric Synthesis - Lecture 1 Background and general principles • Why asymmetric synthesis? The need to prepare pharmaceuticals and other fine chemicals as single enantiomers drives the field of asymmetric synthesis. -

Total Synthesis of Natural Products: a Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65Th Birthday”
molecules Editorial Editorial to the Special Issue “Total Synthesis of Natural Products: A Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65th Birthday” Ari M. P. Koskinen Department of Chemistry and Materials Science, Aalto University School of Chemical Engineering, Kemistintie 1, P.O. Box 16100, 02150 Espoo, Finland; ari.koskinen@aalto.fi Received: 4 December 2020; Accepted: 9 December 2020; Published: 10 December 2020 Natural products have intrigued humans throughout history. Plants with physiological activities, fermentation products, extracts with aroma, scent, or other properties have catalyzed the development of physical methods of separation of compounds and eventually the chemical synthesis of compounds. It is no coincidence that the first synthesis of an organic compound was that of a natural product. Since Wöhler’s synthesis of urea (no stereocenters) nearly two centuries ago, the synthesis of natural products has evolved through Komppa’s synthesis of camphor (one independent stereocenter) a century ago to a stage where compounds of enormous complexity can be attained through chemical synthesis (e.g., palytoxin with 64 stereocenters by Kishi in 1994). This development continues undauntedly, and it stimulates the invention of new synthetic reactions, new technological inventions, and new strategic thinking. The synthesis of natural products also stimulates the minds of medicinal chemists to develop ever better pharmaceutical products inspired by nature. Professor Dieter Schinzer had his initial training in organic synthesis with eminent mentors (Professors Manfred Reetz, Clayton Heathcock and Ekkehard Winterfeldt). Despite his wide research interests in organometallic chemistry (especially silicon, tin and manganese), synthetic methodology and medicinal chemistry, Dr. Schinzer is first and foremost a devoted natural product chemist.