Reaction of Glucose Catalyzed by Framework and Extraframework Tin Sites in Zeolite Beta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
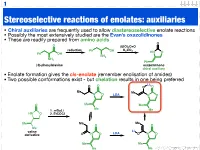
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Total Synthesis of Natural Products: a Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65Th Birthday”
molecules Editorial Editorial to the Special Issue “Total Synthesis of Natural Products: A Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65th Birthday” Ari M. P. Koskinen Department of Chemistry and Materials Science, Aalto University School of Chemical Engineering, Kemistintie 1, P.O. Box 16100, 02150 Espoo, Finland; ari.koskinen@aalto.fi Received: 4 December 2020; Accepted: 9 December 2020; Published: 10 December 2020 Natural products have intrigued humans throughout history. Plants with physiological activities, fermentation products, extracts with aroma, scent, or other properties have catalyzed the development of physical methods of separation of compounds and eventually the chemical synthesis of compounds. It is no coincidence that the first synthesis of an organic compound was that of a natural product. Since Wöhler’s synthesis of urea (no stereocenters) nearly two centuries ago, the synthesis of natural products has evolved through Komppa’s synthesis of camphor (one independent stereocenter) a century ago to a stage where compounds of enormous complexity can be attained through chemical synthesis (e.g., palytoxin with 64 stereocenters by Kishi in 1994). This development continues undauntedly, and it stimulates the invention of new synthetic reactions, new technological inventions, and new strategic thinking. The synthesis of natural products also stimulates the minds of medicinal chemists to develop ever better pharmaceutical products inspired by nature. Professor Dieter Schinzer had his initial training in organic synthesis with eminent mentors (Professors Manfred Reetz, Clayton Heathcock and Ekkehard Winterfeldt). Despite his wide research interests in organometallic chemistry (especially silicon, tin and manganese), synthetic methodology and medicinal chemistry, Dr. Schinzer is first and foremost a devoted natural product chemist. -

Ii- Carbohydrates of Biological Importance
Carbohydrates of Biological Importance 9 II- CARBOHYDRATES OF BIOLOGICAL IMPORTANCE ILOs: By the end of the course, the student should be able to: 1. Define carbohydrates and list their classification. 2. Recognize the structure and functions of monosaccharides. 3. Identify the various chemical and physical properties that distinguish monosaccharides. 4. List the important monosaccharides and their derivatives and point out their importance. 5. List the important disaccharides, recognize their structure and mention their importance. 6. Define glycosides and mention biologically important examples. 7. State examples of homopolysaccharides and describe their structure and functions. 8. Classify glycosaminoglycans, mention their constituents and their biological importance. 9. Define proteoglycans and point out their functions. 10. Differentiate between glycoproteins and proteoglycans. CONTENTS: I. Chemical Nature of Carbohydrates II. Biomedical importance of Carbohydrates III. Monosaccharides - Classification - Forms of Isomerism of monosaccharides. - Importance of monosaccharides. - Monosaccharides derivatives. IV. Disaccharides - Reducing disaccharides. - Non- Reducing disaccharides V. Oligosaccarides. VI. Polysaccarides - Homopolysaccharides - Heteropolysaccharides - Carbohydrates of Biological Importance 10 CARBOHYDRATES OF BIOLOGICAL IMPORTANCE Chemical Nature of Carbohydrates Carbohydrates are polyhydroxyalcohols with an aldehyde or keto group. They are represented with general formulae Cn(H2O)n and hence called hydrates of carbons. -

New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-Yl)-Proline: Efficient, Reusable, and Heterogeneo
molecules Article New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction Omar Sánchez-Antonio 1 , Kevin A. Romero-Sedglach 1 , Erika C. Vázquez-Orta 1 and Eusebio Juaristi 1,2,* 1 Departamento de Química, Centro de Investigación y de Estudios Avanzados, Avenida IPN # 2508, 07360 Ciudad de México, Mexico; [email protected] (O.S.-A.); [email protected] (K.A.R.-S.); [email protected] (E.C.V.-O.) 2 El Colegio Nacional, Luis González Obregón # 23, Centro Histórico, 06020 Ciudad de México, Mexico * Correspondence: [email protected] or [email protected] Academic Editor: Derek J. McPhee Received: 16 September 2020; Accepted: 1 October 2020; Published: 3 October 2020 Abstract: Novel organocatalytic systems based on the recently developed (S)-proline derivative (2S)-[5-(benzylthio)-4-phenyl-(1,2,4-triazol)-3-yl]-pyrrolidine supported on mesoporous silica were prepared and their efficiency was assessed in the asymmetric aldol reaction. These materials were fully characterized by FT-IR, MS, XRD, and SEM microscopy, gathering relevant information regarding composition, morphology, and organocatalyst distribution in the doped silica. Careful optimization of the reaction conditions required for their application as catalysts in asymmetric aldol reactions between ketones and aldehydes afforded the anticipated aldol products with excellent yields and moderate diastereo- and enantioselectivities. The recommended experimental protocol is simple, fast, and efficient providing the enantioenriched aldol product, usually without the need of a special work-up or purification protocol. This approach constitutes a remarkable improvement in the field of heterogeneous (S)-proline-based organocatalysis; in particular, the solid-phase silica-bonded catalytic systems described herein allow for a substantial reduction in solvent usage. -

Structures and Characteristics of Carbohydrates in Diets Fed to Pigs: a Review Diego M
Navarro et al. Journal of Animal Science and Biotechnology (2019) 10:39 https://doi.org/10.1186/s40104-019-0345-6 REVIEW Open Access Structures and characteristics of carbohydrates in diets fed to pigs: a review Diego M. D. L. Navarro1, Jerubella J. Abelilla1 and Hans H. Stein1,2* Abstract The current paper reviews the content and variation of fiber fractions in feed ingredients commonly used in swine diets. Carbohydrates serve as the main source of energy in diets fed to pigs. Carbohydrates may be classified according to their degree of polymerization: monosaccharides, disaccharides, oligosaccharides, and polysaccharides. Digestible carbohydrates include sugars, digestible starch, and glycogen that may be digested by enzymes secreted in the gastrointestinal tract of the pig. Non-digestible carbohydrates, also known as fiber, may be fermented by microbial populations along the gastrointestinal tract to synthesize short-chain fatty acids that may be absorbed and metabolized by the pig. These non-digestible carbohydrates include two disaccharides, oligosaccharides, resistant starch, and non-starch polysaccharides. The concentration and structure of non-digestible carbohydrates in diets fed to pigs depend on the type of feed ingredients that are included in the mixed diet. Cellulose, arabinoxylans, and mixed linked β-(1,3) (1,4)-D-glucans are the main cell wall polysaccharides in cereal grains, but vary in proportion and structure depending on the grain and tissue within the grain. Cell walls of oilseeds, oilseed meals, and pulse crops contain cellulose, pectic polysaccharides, lignin, and xyloglucans. Pulse crops and legumes also contain significant quantities of galacto-oligosaccharides including raffinose, stachyose, and verbascose. -

Extending Enzyme Molecular Recognition with an Expanded Amino Acid Alphabet
Extending enzyme molecular recognition with an expanded amino acid alphabet Claire L. Windlea,b, Katie J. Simmonsa,c, James R. Aulta,b, Chi H. Trinha,b, Adam Nelsona,c,1, Arwen R. Pearsona,2,3, and Alan Berrya,b,1 aAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, United Kingdom; bSchool of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, United Kingdom; and cSchool of Chemistry, University of Leeds, Leeds LS2 9JT, United Kingdom Edited by Perry Allen Frey, University of Wisconsin–Madison, Madison, WI, and approved January 20, 2017 (received for review October 26, 2016) Natural enzymes are constructed from the 20 proteogenic amino for catalysis and arise through posttranslational modifications of acids, which may then require posttranslational modification or the the polypeptide chain (21, 22), allowing access to chemistries not recruitment of coenzymes or metal ions to achieve catalytic function. otherwise provided by the 20 proteogenic amino acids. Here, we demonstrate that expansion of the alphabet of amino acids Technologies for the protein engineer to incorporate Ncas into can also enable the properties of enzymes to be extended. A proteins at specifically chosen sites, either by genetic means (23–25) chemical mutagenesis strategy allowed a wide range of noncanon- or by chemical modification (26, 27), have recently been developed. ical amino acids to be systematically incorporated throughout an These approaches are powerful because, unlike traditional protein active site to alter enzymic substrate specificity. Specifically, 13 engineering with the 20 canonical amino acids, protein engineering different noncanonical side chains were incorporated at 12 different with Ncas has almost unlimited novel side-chain structures and positions within the active site of N-acetylneuraminic acid lyase chemistries from which to choose. -

Tripeptide-Catalyzed Asymmetric Aldol Reaction Between Α-Ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions
catalysts Article Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions Kazumasa Kon 1, Hiromu Takai 2, Yoshihito Kohari 3,* and Miki Murata 1,2,3 1 Graduate School of Manufacturing Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] 2 Graduate School of Materials Science and Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] 3 School of Earth, Energy and Environmental Engineering, Faculty of Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-157-26-9432 Received: 17 April 2019; Accepted: 6 June 2019; Published: 9 June 2019 Abstract: Here, we report the tripeptide-catalyzed asymmetric aldol reaction between α-ketoesters and acetone under acidic cocatalysts-free conditions. H-Pro-Tle-Gly-OH 3g-catalyzed reactions between α-ketoesters and acetone resulted in up to 95% yield and 88% ee. Analysis of the transition state using density functional theory (DFT) calculations revealed that the tert-butyl group in 3g played an important role in enantioselectivity. Keywords: organocatalyst; aldol reaction; peptide catalyst; α-ketoesters 1. Introduction Optically active tertiary alcohols are partial structures present in various natural products and biologically active compounds [1–5]. Various synthetic methods for these compounds have been developed. Asymmetric nucleophile addition to functionalized ketones is one of the most useful synthetic methods, because highly functionalized optically active tertiary alcohols, which can undergo various transformations, can be obtained. -

Conformational Study of the Open-Chain and Furanose Structures of D-Erythrose and D-Threose ⇑ Luis Miguel Azofra A, Ibon Alkorta A, , José Elguero A, Paul L
Carbohydrate Research 358 (2012) 96–105 Contents lists available at SciVerse ScienceDirect Carbohydrate Research journal homepage: www.elsevier.com/locate/carres Conformational study of the open-chain and furanose structures of D-erythrose and D-threose ⇑ Luis Miguel Azofra a, Ibon Alkorta a, , José Elguero a, Paul L. A. Popelier b,c a Instituto de Química Médica, CSIC, Juan de la Cierva, 3, E-28006 Madrid, Spain b Manchester Interdisciplinary Biocentre (MIB), 131 Princess Street, Manchester M1 7DN, United Kingdom c School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, United Kingdom article info abstract Article history: The potential energy surfaces for the different configurations of the D-erythrose and D-threose (open- Received 3 May 2012 chain, a- and b-furanoses) have been studied in order to find the most stable structures in the gas phase. Received in revised form 18 June 2012 For that purpose, a large number of initial structures were explored at B3LYP/6-31G(d) level. All the min- Accepted 19 June 2012 ima obtained at this level were compared and duplicates removed. A further reoptimization of the Available online 27 June 2012 remaining structures was carried out at B3LYP/6-311++G(d,p) level. We characterized 174 and 170 min- ima for the open-chain structures of D-erythrose and D-threose, respectively, with relative energies that Keywords: range over an interval of just over 50 kJ/mol. In the case of the furanose configurations, the number of D-Erythrose minima is smaller by approximately one to two dozen. G3B3 calculations on the most stable minima indi- D-Threose DFT cate that the a-furanose configuration is the most stable for both D-erythrose and D-threose.