Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
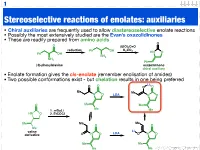
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Studies Toward Synthesis of Polycyclic Polyprenylated Acylphloroglucinols
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Roxana Ciochina University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ciochina, Roxana, "STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS" (2006). University of Kentucky Doctoral Dissertations. 291. https://uknowledge.uky.edu/gradschool_diss/291 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Roxana Ciochina The Graduate School University of Kentucky 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Sciences at the University of Kentucky By Roxana Ciochina Lexington, KY Director: Dr. R. B. Grossman, Professor of Chemistry Lexington, KY 2006 ABSTRACT OF DISSERTATION STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of compounds that reveal intriguing biological activities and interesting and challenging chemical structures. These products are claimed to possess antioxidant, antiviral, and antimitotic properties. Increasing interest is related to their function in the CNS as modulators of neurotransmitters associated to neuronal damaging and depression. All these features make PPAPs targets for synthesis. We decided to focus our own initial efforts in this area on the type A PPAP, nemorosone because we thought that its fairly simple structure relative to other PPAPs would present fewer hurdles as we developed our methodology. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Synthesis of Alkynyl Ribofuranosides
City University of New York (CUNY) CUNY Academic Works Dissertations and Theses City College of New York 2011 Synthesis of Alkynyl Ribofuranosides Christian Rodriguez CUNY City College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/cc_etds_theses/25 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] SYNTHESIS OF ALKYNYL RIBOFURANOSIDES A Thesis Presented to The Faculty of the Chemistry Program The City College of New York In (Partial) Fulfillment of the Requirements for the Degree Master of Arts by Christian Rodriguez December, 2010 - 1 - Synthesis of Alkynyl Ribofuranosides By Christian Rodriguez Mentor: P. Meleties Table of Contents Chapter 1 1.1 Introduction 6 1.2 Preparation of ribonolactone template 8 1.3 Synthesis of protected ribonolactone 9 Chapter 2 2.1 Preparation of ethynyl ribofuranosides 10 2.2 Reaction with ethynylmagnesium bromide 12 2.3 Intramolecular cyclization of diyne diol 14 2.4 Reaction of ribonolactone with lithium acetylide 15 2.5 Boron trifluoride hemiacetal deoxygenation 15 2.6 Alternative deacetylation 16 Chapter 3 3.1 Selecting appropriate protecting group 19 3.2 Preparation of trimethylsilyl alkynyl 5-O-benzyl-2,3-O isopropylidene 19 ribofuranoside 3.3 Lewis acid promoted triethylsilane dehydroxylation mechanism 20 Chapter 4 4.1 Hemiacetal alkynylation 25 4.2 Hemiacetal nucleophilic addition mechanism 26 4.3 Intramolecular -
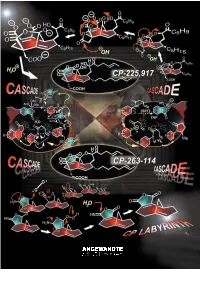
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

Total Synthesis of Natural Products: a Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65Th Birthday”
molecules Editorial Editorial to the Special Issue “Total Synthesis of Natural Products: A Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65th Birthday” Ari M. P. Koskinen Department of Chemistry and Materials Science, Aalto University School of Chemical Engineering, Kemistintie 1, P.O. Box 16100, 02150 Espoo, Finland; ari.koskinen@aalto.fi Received: 4 December 2020; Accepted: 9 December 2020; Published: 10 December 2020 Natural products have intrigued humans throughout history. Plants with physiological activities, fermentation products, extracts with aroma, scent, or other properties have catalyzed the development of physical methods of separation of compounds and eventually the chemical synthesis of compounds. It is no coincidence that the first synthesis of an organic compound was that of a natural product. Since Wöhler’s synthesis of urea (no stereocenters) nearly two centuries ago, the synthesis of natural products has evolved through Komppa’s synthesis of camphor (one independent stereocenter) a century ago to a stage where compounds of enormous complexity can be attained through chemical synthesis (e.g., palytoxin with 64 stereocenters by Kishi in 1994). This development continues undauntedly, and it stimulates the invention of new synthetic reactions, new technological inventions, and new strategic thinking. The synthesis of natural products also stimulates the minds of medicinal chemists to develop ever better pharmaceutical products inspired by nature. Professor Dieter Schinzer had his initial training in organic synthesis with eminent mentors (Professors Manfred Reetz, Clayton Heathcock and Ekkehard Winterfeldt). Despite his wide research interests in organometallic chemistry (especially silicon, tin and manganese), synthetic methodology and medicinal chemistry, Dr. Schinzer is first and foremost a devoted natural product chemist. -

Appendix I: Named Reactions Single-Bond Forming Reactions Co
Appendix I: Named Reactions 235 / 335 432 / 533 synthesis / / synthesis Covered in Covered Featured in problem set problem Single-bond forming reactions Grignard reaction various Radical couplings hirstutene Conjugate addition / Michael reaction strychnine Stork enamine additions Aldol-type reactions (incl. Mukaiyama aldol) various (aldol / Claisen / Knoevenagel / Mannich / Henry etc.) Asymmetric aldol reactions: Evans / Carreira etc. saframycin A Organocatalytic asymmetric aldol saframycin A Pseudoephedrine glycinamide alkylation saframycin A Prins reaction Prins-pinacol reaction problem set # 2 Morita-Baylis-Hillman reaction McMurry condensation Gabriel synthesis problem set #3 Double-bond forming reactions Wittig reaction prostaglandin Horner-Wadsworth-Emmons reaction prostaglandin Still-Gennari olefination general discussion Julia olefination and heteroaryl variants within the Corey-Winter olefination prostaglandin Peterson olefination synthesis Barton extrusion reaction Tebbe olefination / other methylene-forming reactions tetrodotoxin hirstutene / Selenoxide elimination tetrodotoxin Burgess dehydration problem set # 3 Electrocyclic reactions and related transformations Diels-Alder reaction problem set # 1 Asymmetric Diels-Alder reaction prostaglandin Ene reaction problem set # 3 1,3-dipolar cycloadditions various [2,3] sigmatropic rearrangement various Cope rearrangement periplanone Claisen rearrangement hirstutene Oxidations – Also See Handout # 1 Swern-type oxidations (Swern / Moffatt / Parikh-Doering etc. N1999A2 Jones oxidation -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-Yl)-Proline: Efficient, Reusable, and Heterogeneo
molecules Article New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction Omar Sánchez-Antonio 1 , Kevin A. Romero-Sedglach 1 , Erika C. Vázquez-Orta 1 and Eusebio Juaristi 1,2,* 1 Departamento de Química, Centro de Investigación y de Estudios Avanzados, Avenida IPN # 2508, 07360 Ciudad de México, Mexico; [email protected] (O.S.-A.); [email protected] (K.A.R.-S.); [email protected] (E.C.V.-O.) 2 El Colegio Nacional, Luis González Obregón # 23, Centro Histórico, 06020 Ciudad de México, Mexico * Correspondence: [email protected] or [email protected] Academic Editor: Derek J. McPhee Received: 16 September 2020; Accepted: 1 October 2020; Published: 3 October 2020 Abstract: Novel organocatalytic systems based on the recently developed (S)-proline derivative (2S)-[5-(benzylthio)-4-phenyl-(1,2,4-triazol)-3-yl]-pyrrolidine supported on mesoporous silica were prepared and their efficiency was assessed in the asymmetric aldol reaction. These materials were fully characterized by FT-IR, MS, XRD, and SEM microscopy, gathering relevant information regarding composition, morphology, and organocatalyst distribution in the doped silica. Careful optimization of the reaction conditions required for their application as catalysts in asymmetric aldol reactions between ketones and aldehydes afforded the anticipated aldol products with excellent yields and moderate diastereo- and enantioselectivities. The recommended experimental protocol is simple, fast, and efficient providing the enantioenriched aldol product, usually without the need of a special work-up or purification protocol. This approach constitutes a remarkable improvement in the field of heterogeneous (S)-proline-based organocatalysis; in particular, the solid-phase silica-bonded catalytic systems described herein allow for a substantial reduction in solvent usage.