II Reduction Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(+)-Obafluorin, a B-Lactone Antibiotic
Illinois Wesleyan University Digital Commons @ IWU Honors Projects Chemistry 4-26-1996 Pursuit of a Chiral Amino Aldehyde Intermediate in the Synthesis of (+)-Obafluorin, a B-Lactone Antibiotic Jim Cwik '96 Illinois Wesleyan University Follow this and additional works at: https://digitalcommons.iwu.edu/chem_honproj Part of the Chemistry Commons Recommended Citation Cwik '96, Jim, "Pursuit of a Chiral Amino Aldehyde Intermediate in the Synthesis of (+)- Obafluorin, a B-Lactone Antibiotic" (1996). Honors Projects. 15. https://digitalcommons.iwu.edu/chem_honproj/15 This Article is protected by copyright and/or related rights. It has been brought to you by Digital Commons @ IWU with permission from the rights-holder(s). You are free to use this material in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This material has been accepted for inclusion by faculty at Illinois Wesleyan University. For more information, please contact [email protected]. ©Copyright is owned by the author of this document. Pursuit of a Chiral Amino Aldehyde Intermediate in the Synthesis of (+)-Obafluorin, a p-Lactone Antibiotic Jim Cwik Dr. Jeffrey A. Frick, Research Advisor Submitted in Partial Fulfillment of the Requirement for Research Honors in Chemistry and Chemistry 499 Illinois Wesleyan University April 26, 1996 - Table of Contents I. List of Spectral Data , 2 II. Abstract. 3 III. Background 4 IV. Introduction 6 V. -

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

Process for Purification of Ethylene Compound Having Fluorine-Containing Organic Group
Office europeen des brevets (fi) Publication number : 0 506 374 A1 @ EUROPEAN PATENT APPLICATION @ Application number: 92302586.0 @ Int. CI.5: C07C 17/38, C07C 21/18 (g) Date of filing : 25.03.92 (30) Priority : 26.03.91 JP 86090/91 (72) Inventor : Kishita, Hirofumi 3-19-1, Isobe, Annaka-shi Gunma-ken (JP) (43) Date of publication of application : Inventor : Sato, Shinichi 30.09.92 Bulletin 92/40 3-5-5, Isobe, Annaka-shi Gunma-ken (JP) Inventor : Fujii, Hideki (84) Designated Contracting States : 3-12-37, Isobe, Annaka-shi DE FR GB Gunma-ken (JP) Inventor : Matsuda, Takashi 791 ~4 YdndSG Anri3k3~shi @ Applicant : SHIN-ETSU CHEMICAL CO., LTD. Gunma-ken (JP) 6-1, Ohtemachi 2-chome Chiyoda-ku Tokyo (JP) @) Representative : Votier, Sidney David et al CARPMAELS & RANSFORD 43, Bloomsbury Square London WC1A 2RA (GB) (54) Process for purification of ethylene compound having fluorine-containing organic group. (57) A process for purifying an ethylene compound having a fluorine-containing organic group (fluorine- containing ethylene compound) by mixing the fluorine-containing ethylene compound with an alkali metal or alkaline earth metal reducing agent, and subjecting the resulting mixture to irradiation with ultraviolet radiation, followed by washing with water. The purification process ensures effective removal of iodides which are sources of molecular iodine, from the fluorine-containing ethylene compound. < h- CO CO o 10 o Q_ LU Jouve, 18, rue Saint-Denis, 75001 PARIS EP 0 506 374 A1 BACKGROUND OF THE INVENTION 1. Field of the Invention 5 The present invention relates to a process for purifying an ethylene compound having a fluorine-containing organic group, and more particularly to a purification process by which iodine contained as impurity in an ethylene compound having a fluorine-containing organic group can be removed effectively. -
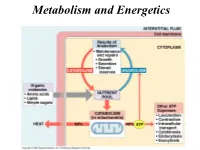
Metabolism and Energetics Oxidation of Carbon Atoms of Glucose Is the Major Source of Energy in Aerobic Metabolism
Metabolism and Energetics Oxidation of carbon atoms of glucose is the major source of energy in aerobic metabolism C6H1206 + 6O2 yields 6 CO2 + H20 + energy Energy released ΔG = - 686 kcal/mol Glucose oxidation requires over 25 discrete steps, with production of 36 ATP. Energy Transformations The mitochondrial synthesis of ATP is not stochiometric. Electron–motive force Proton-motive force Phosphoryl-transfer potential in the form of ATP. Substrate level phosphorylation The formation of ATP by substrate-level phosphorylation ADP ATP P CH O P CH2 O P 2 is used to represent HC OH HC OH a phosphate ester: phosphoglycerate CH OH OH CH2 O P kinase 2 P O bisphosphoglycerate 3-phosphoglycerate OH ADP ATP CH2 CH2 CH3 C O P C OH C O pyruvate kinase non-enzymic COOH COOH COOH phosphoenolpyruvate enolpyruvate pyruvate Why ATP? The reaction of ATP hydrolysis is very favorable ΔGo = - 30.5 kJ/mol = - 7.3 kCal/mol 1. Charge separation of closely packed phosphate groups provides electrostatic relief. Mg2+ 2. Inorganic Pi, the product of the reaction, is immediately resonance-stabilized (electron density spreads equally to all oxygens). 3. ADP immediately ionizes giving H+ into a low [H+] environment (pH~7). 4. Both Pi and ADP are more favorably solvated by water than one ATP molecule. 5. ATP is water soluble. The total body content of ATP and ADP is under 350 mmol – about 10 g, BUT … the amount of ATP synthesized and used each day is about 150 mol – about 110 kg. ATP Production - stage 1 - Glycolysis Glycolysis When rapid production of ATP is needed. -

UNITED STATES PATENT Office PREPARATION of BETA-Akoxy MONO Carboxylcacds Frederick E
Patented July 4, 1944 UNITED STATES PATENT office PREPARATION OF BETA-AKOxY MONO CARBOXYLCACDs Frederick E. King, Akron, Ohio, assignor to The B. F. Goodrich Company, New corporation of New York . York, N. Y., a No Drawing. Application August 5, 1941, Sera No. 405,512 4 Claims. (CL 260-535) This invention relates to a novel process for at least one hydrogen atom on the alpha carbon the preparation of beta-alkoxy derivatives of atom, for example, beta-lactones of saturated monocarboxylic acids, particularly beta-alkoxy aliphatic monocarboxylic acids such as beta-hy derivatives of saturated aliphatic monocarboxylic droxypropionic acid lactone, commonly known as acids such as beta-alkoxy proponic acids, and to 5 hydracrylic acid lactone, beta-hydroxy butyric the conversion of such acids into alkyl esters of acid lactone, alpha-methyl hydracrylic acid lac alpha beta unsaturated monocarboxylic acids tone, beta-hydroxy n-valeric acid actone, beta such as the alkyl esters of acrylic and meth hydroxy alpha-methyl butyric acid lactone, al acrylic acids. - - pharethyl hydracrylic acid lactone, beta-hydroxy In a copending application Serial No. 393,671, 0 isovaleric acid lactone, beta-hydroxy n-caproic filed May 15, 1941, an economical method of pre acid lactone, beta-hydroxy alpha-methyl valeric paring lactones of beta-hydroxy Carboxylic acids acid lactone, beta-methyl beta-ethyl hydracrylic from the reaction of a ketene with a carbonyl acid lactone, alpha-methyl beta-ethylhydracrylic compound such as an aldehyde or ketone has acid lactone, alpha-propyl -

The First General Electron Transfer Reductions of Carboxylic Acid Derivatives Using Samarium Diiodide
The First General Electron Transfer Reductions of Carboxylic Acid Derivatives Using Samarium Diiodide A thesis submitted to The University of Manchester for the degree of Doctor of Philosophy In the faculty of Engineering and Physical Sciences 2014 Malcolm Peter Spain School of Chemistry Malcolm Spain PhD Thesis Contents Abstract .................................................................................................................... 5 Declaration................................................................................................................ 6 Copyright statement .................................................................................................. 7 Acknowledgements ................................................................................................... 9 Abbreviations .......................................................................................................... 10 Chapter 1. Introduction ........................................................................................... 13 1.1 Introduction to samarium diiodide .................................................................. 13 1.2 Reduction of ketones and aldehydes ............................................................. 15 1.3 Reduction of carboxylic acid derivatives ....................................................... 23 Chapter 2. Investigations of the SmI2–H2O system ................................................. 27 2.0 Preliminary studies of the SmI2–H2O system ................................................ -

Studies Toward Synthesis of Polycyclic Polyprenylated Acylphloroglucinols
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Roxana Ciochina University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Ciochina, Roxana, "STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS" (2006). University of Kentucky Doctoral Dissertations. 291. https://uknowledge.uky.edu/gradschool_diss/291 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Roxana Ciochina The Graduate School University of Kentucky 2006 STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Arts and Sciences at the University of Kentucky By Roxana Ciochina Lexington, KY Director: Dr. R. B. Grossman, Professor of Chemistry Lexington, KY 2006 ABSTRACT OF DISSERTATION STUDIES TOWARD SYNTHESIS OF POLYCYCLIC POLYPRENYLATED ACYLPHLOROGLUCINOLS Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of compounds that reveal intriguing biological activities and interesting and challenging chemical structures. These products are claimed to possess antioxidant, antiviral, and antimitotic properties. Increasing interest is related to their function in the CNS as modulators of neurotransmitters associated to neuronal damaging and depression. All these features make PPAPs targets for synthesis. We decided to focus our own initial efforts in this area on the type A PPAP, nemorosone because we thought that its fairly simple structure relative to other PPAPs would present fewer hurdles as we developed our methodology. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

CHE202 Reductions and Heterocycles
CHE202 Structure & Reactivity in Organic Chemistry: ! Reduction Reactions and Heterocyclic Chemistry! 9 lectures, Semester B 2014! Dr. Chris Jones! ! [email protected]! Office: 1.07 Joseph Priestley Building! ! Office hours:! 9.30-10.30 am Tuesday! 1.30-2.30 pm Thursday (by appointment only)! Course structure and recommended texts! 2! §" Coursework:! !Semester B – week 9 ! !5% (‘Coursework 7’)! !Semester B – week 11! !5% (‘Coursework 8’)! ! §" Test:! !Semester B – week 12! !15% (‘Test 4’)! §" Recommended text books:! ‘Organic Chemistry’, Clayden, ‘Oxidation & Reduction in ‘Heterocyclic Chemistry’, Greeves & Warren, OUP, 2012.! Organic Chemistry’, Donohoe, Joule & Mills, Wiley, 2010.! OUP, 2000.! Don’t forget clickers! Overview of Reduction Chemistry lecture material! 3! §" Reduction:! - Definition (recap.)! - Reduction of carbon-carbon double and triple bonds! - Heterogeneous hydrogenation! - Homogeneous hydrogenation, including stereoselective hydrogenation! - Dissolved metal reductions! - Other methods of reduction! - Reduction of carbon-heteroatom double and triple bonds! - Reduction of carbonyl derivatives, addressing chemoselectivity! - Stereoselective reduction of carbonyl derivatives! - Reduction of imines and nitriles! - Reductive cleavage reactions! - Hydrogenolysis of benzyl and allyl groups! - Dissolved metal reduction! - Deoxygenation reactions! - Reduction of heteroatom functional groups! e.g. azides, nitro groups, N-O bond cleavage! Reduction: definition! 4! §" Reduction of an organic substrate can be defined as:! - The concerted -

Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction
Detection of Phenethylamine, Amphetamine, and Tryptamine Imine By-Products from an Acetone Extraction Mary A. Yohannan* and Arthur Berrier U.S. Department of Justice Drug Enforcement Administration Special Testing and Research Laboratory 22624 Dulles Summit Court Dulles, VA 20166 [email: mary.a.yohannan -at- usdoj.gov] ABSTRACT: The formation of imine by-products from phenethylamines, amphetamines, and tryptamines upon an acetone extraction is presented. These imine by-products were characterized using GC/MSD and exhibited preferential cleavage at the α-carbon of the alkyl chain. Further characterization of the imine by-products of phenethylamine and tryptamine was done using IR and NMR. KEYWORDS: phenethylamine, tryptamine, imine, acetone, schiff base, drug chemistry, forensic chemistry In most forensic laboratories, the solvents used to extract at the α-carbon on the alkyl chain. In addition to GC/MS, the drugs are chosen based upon their solubility properties and their imines formed from phenethylamine base and tryptamine base ability to not interact with the drug. In fact, there are very few were characterized by Fourier transform-infrared spectroscopy publications where a solvent used to extract a drug reacts with (FTIR) and nuclear magnetic resonance (NMR) spectroscopy. the drug and forms by-products [1-3]. This laboratory recently discovered that an additional Experimental component was formed when acetone was used to extract a Solvents, Chemicals, and Materials sample containing a known tryptamine. Analysis by gas Acetone was ACS/HPLC grade from Burdick and Jackson chromatography/mass spectroscopy (GC/MS) of the acetone Laboratories (Muskegon, MI). Phenethylamine base and extract yielded an extra peak in the total ion chromatogram that tryptamine base were obtained from Sigma-Aldrich Chemicals was approximately half the abundance of the known tryptamine (Milwaukee, WI). -

Selective Deoxygenation of Biomass‐Derived Bio‐Oils Within Hydrogen‐Modest Environments
Edinburgh Research Explorer Selective Deoxygenation of Biomass-Derived Bio-oils within Hydrogen-Modest Environments: A Review and New Insights Citation for published version: Rogers, K & Zheng, Y 2016, 'Selective Deoxygenation of Biomass-Derived Bio-oils within Hydrogen-Modest Environments: A Review and New Insights', Chemsuschem, vol. 9, DOI:10.1002/cssc.201600144, pp. 1750- 1772. <http://onlinelibrary.wiley.com/doi/10.1002/cssc.201600144/abstract> Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Chemsuschem General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 07. Oct. 2021 DOI:10.1002/cssc.201600144 Reviews Selective Deoxygenation of Biomass-Derived Bio-oils within Hydrogen-Modest Environments:AReview and New Insights Kyle A. Rogers[a] and Ying Zheng*[a, b] ChemSusChem 2016, 9,1750 –1772 1750 2016 The Authors. PublishedbyWiley-VCH Verlag GmbH &Co. KGaA, Weinheim Reviews Research development of processes for refiningbio-oils is be- ing acombination of oxophilicity and an active metal phase comingincreasingly popular.One issue that these processes appear to be the most beneficial for selectivedeoxygenation possess is their high requirement for H2 gas.