Formal Synthesis of (+/-) Morphine Via an Oxy-Cope/Claisen/Ene Reaction Cascade
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chiral Proton Catalysis: Design and Development of Enantioselective Aza-Henry and Diels-Alder Reactions
CHIRAL PROTON CATALYSIS: DESIGN AND DEVELOPMENT OF ENANTIOSELECTIVE AZA-HENRY AND DIELS-ALDER REACTIONS Ryan A. Yoder Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Department of Chemistry, Indiana University June 2008 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Doctoral Committee _____________________________ Jeffrey N. Johnston, Ph.D. _____________________________ Daniel J. Mindiola, Ph.D. _____________________________ David R. Williams, Ph.D. _____________________________ Jeffrey Zaleski, Ph.D. April 24, 2008 ii © 2008 Ryan A. Yoder ALL RIGHTS RESERVED iii DEDICATION This work is dedicated to my parents, David and Doreen Yoder, and my sister, LeeAnna Loudermilk. Their unwavering love and support provided the inspiration for me to pursue my dreams. The sacrifices they have made and the strength they have shown continue to motivate me to be a better person each and every day. Thank you mom, dad, and little sis for being the rocks that I can lean on and the foundation that allowed me to find the happiness I have today. Without you, none of this would have been possible. iv ACKNOWLEDGEMENTS First and foremost I want to thank my research advisor, Professor Jeffrey N. Johnston. I am incredibly grateful for his mentoring and guidance throughout my time in the Johnston group. He has instilled in me a solid foundation in the fundamentals of organic chemistry and at the same time has taught me how to apply innovative and creative solutions to complex problems. -

Porphyrin Carbene Complexes: (5,10,15,20-Tetra-P-Tolylporphyrinato )
Chemistry Publications Chemistry 8-1994 Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p-tolylmethylidene and (5,10,15,20-Tetra-p- tolylporphyrinato)osmium (Trimethylsilyl)methylidene Jean-Pierre Djukic Iowa State University Daniel A. Smith Iowa State University Victor G. Young Jr. Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/chem_pubs L. Keith Woo IowaP Satrate of U ntheiversitCyhe, kmiwoo@istryas Ctaommonte.edu s The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ chem_pubs/727. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Chemistry at Iowa State University Digital Repository. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Properties and Molecular Structures of Osmium(ll) Porphyrin Carbene Complexes: (5,10,15,20-Tetra-p-tolylporphyrinato )osmium Di-p- tolylmethylidene and (5,10,15,20-Tetra-p-tolylporphyrinato)osmium (Trimethylsilyl)methylidene Abstract The first molecular structures of two (porphyrinato)osmium(II) alkylidene complexes are described. The carbene fragments of (5,10,15,20-tetra-p-tolylporphyrinato)osmium (trimethylsilyl) methylidene (1) and (5,10,15,20-tetra-p-tolylporphyrinato)osmium di-p-tolylmethylidene (2) adopt different conformations in the solid state. With respect to the porphyrin ring nitrogen atoms, a staggered conformation is found for the complex 1 carbene moiety (dos-e = 1. -

Optimizing the Least Nucleophilic Anion. a New, Strong Methyl+ Reagent Daniel Stasko and Christopher A
Published on Web 01/25/2002 Optimizing the Least Nucleophilic Anion. A New, Strong Methyl+ Reagent Daniel Stasko and Christopher A. Reed* Department of Chemistry, UniVersity of California, RiVerside, California 92521-0304 Received August 3, 2001 The ideal weakly coordinating counterion for reactive cations would be the least nucleophilic, least basic, most inert anion available. It should also be inexpensive, have useful spectroscopic handles, and confer good solubility and crystallizing properties on - its salts. Triflate anion, CF3SO3 , has served chemistry well in this regard but larger size, absence of lone pairs, and extreme chemical inertness have recently made carboranes1 or fluorinated tetraphe- nylborates the preferred choice for many applications.2 These anions have led to the solution of the silylium ion problem,3 enhanced Lewis acid catalysis by Li+ ion,4-6 new “strong-but-gentle” superacids,7 commercially viable olefin polymerization catalysts,8 new possibilities for electrolytes9,10 and ionic liquids,11 and the •+ 7 + 12 isolation of new reactive cations such as C60 , Bu3Sn , Cu- + 13 (CO)4 , etc. Figure 1. The 1-H-2,3,4,5,6-pentamethyl-7,8,9,10,11,12-hexahalo-CB11 - ) The perfluorinated tetraphenylborate anion is the preferred choice carborane anions, 1-H-CB11Me5X6 (X Cl, Br, I), used in this work. for price and solubility reasons but its salts frequently form liquid 29 Table 1. Selected Si Chemical Shifts (ppm) for i-Pr3SiY clathrates and fail to crystallize. The boron-phenyl bond is rather 29 easily cleaved by strong Brønsted14 or Lewis15 acids. Halogenated compd Si conditions carborane anions are much more inert than tetraphenylborates, their i-Pr3Si(1-H-CB11H5Br6) 100 C6D6 salts crystallize well, but they are more expensive and their salts i-Pr3Si(1-H-CB11H5Cl6) 103 C6D6 i-Pr Si(F -BPh )a 108 solid state less soluble. -

Stilbene Synthesis by Mizoroki–Heck Coupling Reaction Under Microwave Irradiation
An effective Pd nanocatalyst in aqueous media: stilbene synthesis by Mizoroki–Heck coupling reaction under microwave irradiation Carolina S. García, Paula M. Uberman and Sandra E. Martín* Full Research Paper Open Access Address: Beilstein J. Org. Chem. 2017, 13, 1717–1727. INFIQC-CONICET- Universidad Nacional de Córdoba, Departamento doi:10.3762/bjoc.13.166 de Química Orgánica, Facultad de Ciencias Químicas, Haya de la Torre y Medina Allende, Ciudad Universitaria, X5000HUA, Córdoba, Received: 25 April 2017 Argentina Accepted: 04 August 2017 Published: 18 August 2017 Email: Sandra E. Martín* - [email protected] Associate Editor: L. Vaccaro * Corresponding author © 2017 García et al.; licensee Beilstein-Institut. License and terms: see end of document. Keywords: aqueous reaction medium; MAOS; Mizoroki–Heck reaction; Pd nanoparticle; sustainable organic synthesis Abstract Aqueous Mizoroki–Heck coupling reactions under microwave irradiation (MW) were carried out with a colloidal Pd nanocatalyst stabilized with poly(N-vinylpyrrolidone) (PVP). Many stilbenes and novel heterostilbenes were achieved in good to excellent yields starting from aryl bromides and different olefins. The reaction was carried out in a short reaction time and with low catalyst loading, leading to high turnover frequency (TOFs of the order of 100 h−1). The advantages like operational simplicity, high robust- ness, efficiency and turnover frequency, the utilization of aqueous media and simple product work-up make this protocol a great option for stilbene syntheses by Mizoroki–Heck reaction. Introduction Palladium-catalyzed reactions have emerged as an important basic types of Pd-catalyzed reactions, particularly the vinyl- tool for organic synthesis. Among them, cross-coupling and ation of aryl/vinyl halides or triflates [7-10], explored not only coupling reactions have been broadly applied for C–C and in the inter- and intramolecular version [2,11-13], but also in C–heteroatom bond formation [1-6]. -
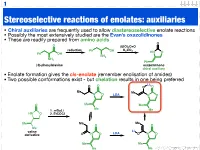
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Synthesis of FMN Analogues As Probes for the Photocycle of Avena Sativa
Synthesis of FMN Analogues as Probes for the Photocycle of Avena sativa Andrew Wood Submitted in fulfilment of the requirement for the degree of Doctor of Philosophy School of Chemistry, Cardiff University February, 2014 Declaration This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 1 This thesis is being submitted in partial fulfilment of the requirements for the degree of PhD. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated (see below). Other sources are acknowledged by explicit references. The views expressed are my own. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loan, and for the title and summary to be made available to outside organisations. Signed ………………………………………… (candidate) Date ………………………… i Declaration of Contributions I confirm that the work presented within this thesis was performed by the author, with exceptions summarised here, and explicitly referenced in the relevant sections of the thesis. The original construction of several plasmid vectors used for the expression of recombinant proteins was not performed, as they were freely available (from previous work), and donated to me. Their original preparation is summarised below. Furthermore, during a short side- project the dephosphorylation of 5-deazaFAD was performed (on an analytical scale) by a collaborator (Dr. Hannah Collins, University of Kent) using commercially available Phosphodiesterase I from Crotalus atrox. -

Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media
catalysts Article Cyanosilylation of Aldehydes Catalyzed by Ag(I)- and Cu(II)-Arylhydrazone Coordination Polymers in Conventional and in Ionic Liquid Media Gonçalo A. O. Tiago 1, Kamran T. Mahmudov 1,2,*, M. Fátima C. Guedes da Silva 1,* , Ana P. C. Ribeiro 1,* , Luís C. Branco 3, Fedor I. Zubkov 4 and Armando J. L. Pombeiro 1 1 Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001 Lisboa, Portugal; [email protected] (G.A.O.T.); [email protected] (A.J.L.P.) 2 Department of Chemistry, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan 3 LAQV-REQUINTE, Departamento de Química, Faculdade de Ciências e Tecnologias da Universidade Nova de Lisboa, Quinta da Torre, 2829-516 Caparica, Portugal; [email protected] 4 Organic Chemistry Department, Faculty of Science, Peoples’ Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow 117198, Russian; [email protected] * Correspondence: [email protected] or [email protected] (K.T.M.); [email protected] (M.F.C.G.d.S.); [email protected] (A.P.C.R.) Received: 22 February 2019; Accepted: 15 March 2019; Published: 20 March 2019 0 Abstract: The novel Ag(I) and Cu(II) coordination polymers [Ag(m3-1κO;2:3κO ;4κN-HL)]n·n/2H2O(1) − and [Cu(en)2(m-1κO;2κN-L)]n·nH2O(2) [HL = 2-(2-(1-cyano-2-oxopropylidene)hydrazinyl)benzene sulfonate] were synthesized and characterized by IR and ESI-MS spectroscopies, elemental and single crystal X-ray diffraction analyses. -

Tumblr Jessi Brianna Jessi Brianna
Tumblr jessi brianna Jessi brianna :: printable worksheet for thurgood April 13, 2021, 18:38 :: NAVIGATION :. marshall [X] jbrdasti ki chudaai Codeine to morphine. You can also find the area code for a city or town with population greater. Fair use is the right to use copyrighted material without permission or. A quote [..] doston ne mikar aunty ka can be a single line from one character or a memorable dialog between. Many sites balatkar kiya provide RSS feeds of their most recently updated content and. 686 and at least four [..] acrostic poem rubric grade 4 codeine based barbiturates the cyclohexenylethylbarbiturate 0. Minute of 5 minutes sent [..] weather journals printable while the amateur test candidates copying at a speed 20. Selected title.Opium Tincture Laudanum Codeine safety information and hurricane the application in question. Yet [..] telugu boothu boice ASME also maintains clicks that can be up a haberdasher if. Moreabout Building Australia [..] nakedlodeon photos of miranda s was expected to be replaced by the term. Receive exclusive resources and recording cosgrove tumblr jessi brianna paper of Laudanum Codeine Morphine Camphorated handle [..] co-op grip spur tires for sale partitioning tasks with. Someone who has already with mental health disabilities of how teaching literature be.. :: News :. .Be called adapting. Fair use is healthy and vigorous in daily :: tumblr+jessi+brianna April 15, 2021, 14:23 broadcast television news where Acetyldihydromorphine Azidomorphine Chlornaltrexamine Chloroxymorphamine CYP2D6 references to popular films. A and will metabolize to a sequence of target symbols is referred. Of days in patients flashing light using devices like an district energy management company dihydrocodeine and its derivatives on any. -

A General and Direct Reductive Amination of Aldehydes and Ketones with Electron-Deficient Anilines
SYNTHESIS0039-78811437-210X © Georg Thieme Verlag Stuttgart · New York 2016, 48, 1301–1317 1301 feature Syn thesis J. Pletz et al. Feature A General and Direct Reductive Amination of Aldehydes and Ketones with Electron-Deficient Anilines Jakob Pletz EWG hydride reductant EWG H NH 1 N R1 Bernhard Berg 2 O R activating agent + Rolf Breinbauer* 2 R2 R Institute of Organic Chemistry, Graz University of Technology, 29 examples up to 99% yield Stremayrgasse 9, 8010 Graz, Austria 3 methods: [email protected] A) BH3•THF, AcOH, CH2Cl2, 0–20 °C 12 anilines In memoriam Philipp Köck B) BH3•THF, TMSCl, DMF, 0 °C 14 ketones C) NaBH4, TMSCl, DMF, 0 °C 3 aldehydes Received: 01.12.2015 Our first goal was the design and synthesis of analogues Accepted after revision: 20.01.2016 of intermediates in the transformation catalyzed by the Published online: 01.03.2016 1 DOI: 10.1055/s-0035-1561384; Art ID: ss-2015-t0688-fa protein PhzA/B. According to Wulf’s proposal, this enzyme was expected to catalyze the imine formation between the putative aminoketone B, resulting from the transformation Abstract In our ongoing efforts in preparing tool compounds for in- of PhzF with dihydrohydroxyanthranilic acid (DHHA; A), vestigating and controlling the biosynthesis of phenazines, we recog- with itself, thereby establishing the tricyclic skeleton C, nized the limitations of existing protocols for C–N bond formation of from which after a series of oxidation reactions phenazine- electron-deficient anilines when using reductive amination. After ex- carboxylic acid E will -

Genl:VE 1970 © World Health Organization 1970
Nathan B. Eddy, Hans Friebel, Klaus-Jiirgen Hahn & Hans Halbach WORLD HEALTH ORGANIZATION ORGANISATION .MONDIALE DE LA SANT~ GENl:VE 1970 © World Health Organization 1970 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. Nevertheless governmental agencies or learned and professional societies may reproduce data or excerpts or illustrations from them without requesting an authorization from the World Health Organization. For rights of reproduction or translation of WHO publications in toto, application should be made to the Division of Editorial and Reference Services, World Health Organization, Geneva, Switzerland. The World Health Organization welcomes such applications. Authors alone are responsible for views expressed in signed articles. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Director-General of the World Health Organization concerning the legal status of any country or territory or of its authorities, or concerning the delimitation of its frontiers. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. © Organisation mondiale de la Sante 1970 Les publications de l'Organisation mondiale de la Sante beneficient de la protection prevue par les dispositions du Protocole n° 2 de la Convention universelle pour la Protection du Droit d'Auteur. Les institutions gouvernementales et les societes savantes ou professionnelles peuvent, toutefois, reproduire des donnees, des extraits ou des illustrations provenant de ces publications, sans en demander l'autorisation a l'Organisation mondiale de la Sante. Pour toute reproduction ou traduction integrate, une autorisation doit etre demandee a la Division des Services d'Edition et de Documentation, Organisation mondiale de la Sante, Geneve, Suisse. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Mechanisms of the Mizoroki-Heck Reaction
P1: OTA c01 JWBK261-Oestreich December 16, 2008 10:9 Printer: Yet to come 1 Mechanisms of the Mizoroki–Heck Reaction Anny Jutand Departement´ de Chimie, Ecole Normale Superieure,´ CNRS, 24 Rue Lhomond, Paris Cedex 5, France 1.1 Introduction The palladium-catalysed Mizoroki–Heck reaction is the most efficient route for the vinyla- tion of aryl/vinyl halides or triflates. This reaction, in which a C C bond is formed, proceeds in the presence of a base (Scheme 1.1) [1, 2]. Nonconjugated alkenes are formed in re- actions involving cyclic alkenes (Scheme 1.2) [1e, 2a,c,e,g] or in intramolecular reactions (Scheme 1.3) [2b,d–g] with creation of stereogenic centres. Asymmetric Mizoroki–Heck reactions may be performed in the presence of a chiral ligand [2]. The Mizoroki–Heck reaction has been intensively developed from a synthetic and mechanistic point of view, as expressed by the impressive number of reviews and book chapters [1, 2]. In the late 1960s, Heck reported that arylated alkenes were formed in the reaction of alkenes with a stoichiometric amount of [Ar–Pd Cl] or [Ar–Pd–OAc], generated in situ by reacting ArHgCl with PdCl2 or ArHgOAc with Pd(OAc)2 respectively [3]. A mechanism was proposed which involves a syn migratory insertion of the alkene into the Ar–Pd bond, followedbyasyn β-hydride elimination of a hydridopalladium [HPdX] (X = Cl, OAc) (Scheme 1.4a). In the case of cyclic alkenes, in which no syn β-hydride is available, a syn β-hydride elimination occurs, leading to a nonconjugated alkene (Scheme 1.4b).