Paper : HCHE4CC09LTH
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
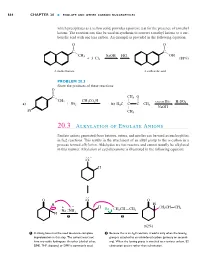
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -
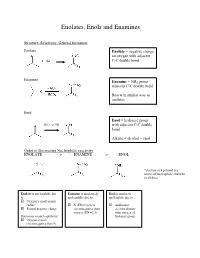
Enolates, Enols and Enamines
Enolates, Enols and Enamines Structure, Reactivity, General formation Enolate Enolate = negative charge on oxygen with adjacent C-C double bond Enamine Enamine = NR2 group adjacent C-C double bond Reacts in similar way as enolates Enol Enol = hydroxyl group - H3O+ or OH with adjacent C-C double bond Alkene + alcohol = enol Order of Decreasing Nucleophilic reactivity ENOLATE > ENAMINE > ENOL *electron-rich pi bond is a source of nucleophilic character in all three Enolate is nucleophilic due Enamine is moderately Enol is moderate to… nucleophilic due to… nucleophile due to… Oxygen’s small atomic radius N (EN=3) is less Additional Formal negative charge electronegative than electron density oxygen (EN =3.5) from oxygen of Detractors to nucleophilicity: hydroxyl group Oxygen is more electronegative than N When looking at which side is favored (products or reactants): Equilibrium favors side with WEAKER acid/base pair Weakest acid and weakest base should be on SAME side When comparing pKa values, juxtapose just the acids or just the bases Side with larger pka value (weaker acid) favored in equilibrium Remember that differences in pKa values reflect a difference quantity of 10x Example: compound H-A (pKa=3) + base compound A (pKa=10) + H-base 7 equilibrium lies to the RIGHT by factor of 10 Some Useful pKa values (from more acidic basic) H2SO4 (pka = -9) H2O (pka = 15.7) + H3O (pka = -1.8) CH3COCH3 (pka = 19) B-diketone (pka = 9) CH3COOCH3 (pKa = 25) HCN (pka = 9.1) LDA-H (pka = 36) CH3OH (pka = 15.5) How do we know which proton to deprotonate? Deprotonate most acidic proton usually leads to the most stable enolate 1. -

Chap 11. Carbonyl Alpha-Substitution Reactions and Condensation Reactions
Chap 11. Carbonyl Alpha-Substitution Reactions and Condensation Reactions Four fundamental reactions of carbonyl compounds 1) Nucleophilic addition (aldehydes and ketones) 2) Nucleophilic acyl substitution (carboxylic acid derivatives) 3) The alpha-substitution Reactions occur at the position next to the carbonyl group–the alpha position–and result in the substitution of an α hydrogen atom by some other electrophilic group, E+: O O E+ α H E + H+ 4) The carbonyl condensation Reactions take place when two carbonyl compounds react with each other in such a way that the α carbon of one partner becomes bonded to the carbonyl carbon of the second partner. O H O OH 2 H The key feature of the reactions (3, 4) is that they take place through the formation of either enol or enolate ion intermediates. H O O C C C C 11.1 Keto-Enol Tautomerism The interconversion between the keto and enol forms of carbonyl compounds is a special kind of isomerism called tautomerism. In Greek, tauto means “the same,” and meros means “part.” The interconversion of tautomers involves the movement of atoms (H), so they are different from resonance forms. H O Rapid O α H Keto tautomer Enol tautomer 1 Most carbonyl compounds exist almost entirely in the keto form at equilibrium. OOHO OH 99.9999% 0.0001% 99.999999% 0.000001% Mechanism of keto-enol tautomerism Acid catalysis H A H H H O O O A O H H H + HA Keto tautomer Enol tautomer Base catalysis HOH H OOOH OO H + OH Keto tautomer Enol tautomer 11.2 Reactivity of Enols: The Mechanism of Alpha-Substitution Reactions Since their double bonds are electron-rich, enols behave as nucleophiles and react with electrophiles in the same way as alkenes do. -

Enolate Chemistry
Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 1 Enolate Chemistry 1. Some Basics In most cases the equilibrium lies almost completely on the side of the ketone. The ketone tautomer is electrophilic and reacts with nucleophiles: The enol tautomer is nucleophilic and reacts with electrophiles. There are two possible products - enols are ambident nucleophiles: The nucleophilic enol tautomer (and especially the enolate variant) is one of the most important reactive species for C-C bond formation. Treat a ketone with an appropriate base and can get deprotonation at the α-position to form an enolate: Enolates are synthetically much more useful than enols (although they react analogously). Imine anions and eneamines are synthetic equivalents of enolate anions. Li N N H H Imine anion eneamine By knowing the pKa values of the relevant acidic protons it is possible to predict suitable bases for forming the corresponding enolates. Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 2 Some Important pKa Values - - - O O HC N HC3 NO2 H2CN+ 2 + O O pKa ~ 10 - Enolates are nucleophiles and ketones are electrophiles - therefore there is always the potential problem for self condensation. If this is desirable we need to use a base which does not completely deprotonate the carbonyl compound i.e. set up an equilibrium. This is best achieved when the pKa of the carbonyl group and conjugate acid (of the base) are similar: Prof. Dr. Burkhard König, Institut für Organische Chemie, Uni Regensburg 3 pKa (tBuOH) ~ 18. A stronger base. pKa difference of 2. Ratio of ketone to enolate will be of the order 100:1 i.e. -

The Α-Carbon Atom and Its Pka the Inductive Effect of the Carbonyl
Chapter 18: Enols and Enolates O E+ O H Enolate O E carbonyl H O E+ Enol 18.1: The α-Carbon Atom and its pKa O "' " #' !' ! # 126 The inductive effect of the carbonyl causes the α-protons to be more acidic. The negative charge of the enolate ion (the conjugate base of the aldehyde or ketone) is stabilized by resonance delocalization. The pKa of the α-protons of aldehydes and ketones is in the range of 16-20 (Table 18.1, p. 754) resonance effect ! - inductive O O !+ O + B effect C H C C + H-B !+ C C C carbonyl Enolate anion H C C C C C C O O O O H3C CH3 C H3CH2COH H3C CH3 ethane acetone ethanol 127 pKa= 50-60 pKa= 19 pKa= 16 65 O O O O O C C Cl C C C H3C CH3 H3C C H3C C H3C C CH3 H H H H H H pKa 19 14 16 9 O O + H O C + H3O C 2 H C CH H3C CH3 3 2 acid base conjugate conjugate base acid pKa 19 -1.7 (weaker acid) (weaker base) (stronger base) (stronger acid) O O + HO C + H O C H C CH 2 H3C CH3 3 2 acid base conjugate conjugate base acid pKa 19 15.7 (weaker acid) (weaker base) (stronger base) (stronger acid) 128 O O O O C C + H2O C C + HO H C C CH H C C CH 3 3 3 3 H H H acid base conjugate conjugate base acid pKa 9 15.7 (stronger acid) (stronger base) (weaker base) (weaker acid) 18.2: The Aldol Condensation- An enolate of one carbonyl (nucleophile) reacts with the carbonyl carbon (electrophile) of a second carbonyl compound resulting in the formation of a new C-C bond O OH O 2 H C C H + HO + HO 3 H3C CH CH2 C H acetaldehyde 3-hydroxybutanal (!-hydroxy aldehyde) 129 66 Mechanism of the base-catalyzed aldol condensation (Fig. -

Chem 353: Aldol Condensation
ALDOL.1 ORGANIC SYNTHESIS: ALDOL CONDENSATION REACTION TECHNIQUES REQUIRED: Filtration (Vacuum), Recrystallisation, Melting Point Determination, Yield calculation OTHER DOCUMENTS: Experimental Procedure, Report template (pdf), Report template (doc), Spectra INTRODUCTION In an "aldol addition" reaction, an enol or more commonly an enolate of an aldehyde or ketone reacts with a second aldehyde or ketone forming a new carbon-carbon bond. This makes the aldol reaction an important reaction for organic synthesis. Originally, the aldol reaction used ethanal (see below) and therefore the product contained both an aldehyde and an alcohol functional group; thus it became known as the aldol reaction. Recall that aldehydes and ketones typically undergo nucleophilic addition reactions. The aldol reaction demonstrates how carbonyl compounds can react as electrophiles or nucleophiles depending on the reaction conditions and what other species are present. Alcohols are similar in that respect. Can you think of examples to illustrate that ? The aldol reaction requires an aldehyde or ketone that contains at least one -hydrogen (the -hydrogen is on the carbon adjacent to the C=O group) since the -hydrogen is required in order to form the enol or enolate. In the base-catalysed aldol reaction, the relatively acidic hydrogen on the -carbon (typical pKa 16-20) is deprotonated by a base to form the enolate. The enolate reacts as a carbon nucleophile that can then react with the electrophilic carbonyl carbon of another aldehyde or ketone molecule. Depending on the strength of the base used, the extent of deprotonation can be controlled. If a strong base is used (such as lithium diisopropylamide, LDA) then deprotonation is quantitative (100%). -

22.7 Alkylation of Ester Enolate Ions
22_BRCLoudon_pgs4-4.qxd 11/26/08 12:27 PM Page 1084 1084 CHAPTER 22 • THE CHEMISTRY OF ENOLATE IONS, ENOLS, AND a,b-UNSATURATED CARBONYL COMPOUNDS O S O S OH C N NH _O acetyl-CoA carboxylase H2CCA" SCoA H H enol form of Lacetyl-CoA+ R S H carboxybiotin O S O O S S HN NH _OCC CH2 SCoA H H LLmalonyl-CoALL + R S H biotin Provide a curved-arrow mechanism for this reaction, using B as a base (which is part of the 3 enzyme) and |BH as its conjugate acid. 22.7 ALKYLATION OF ESTER ENOLATE IONS Sections 22.4–22.6 described reactions in which enolate ions react as nucleophiles at the car- bonyl carbon atom. This section considers two reactions in which enolate ions are used as nu- cleophiles in SN2 reactions. A. Malonic Ester Synthesis Diethyl malonate (malonic ester), like many other b-dicarbonyl compounds, has unusually acidic a-hydrogens. (Why?) Consequently, its conjugate-base enolate ion can be formed nearly completely with alkoxide bases such as sodium ethoxide. O O O O S S S S _ (22.64a) EtO_ EtOC CH2 C OEt EtO H EtOC CH C OEt 2 3 ++LL LL 2 L enolateLL ion of 2diethylLL malonate 2 diethyl malonate 2 pKa 12.9 = The conjugate-base anion of diethyl malonate is nucleophilic, and it reacts with alkyl halides and sulfonate esters in typical SN2 reactions. Such reactions can be used to introduce alkyl groups at the a-position of malonic ester. CH2CH3 CH2CH3 Na CH(CO Et) CH "CH Br CH "CHCH(CO Et) Na Br (22.64b) | _ 2 2 3 EtOH 3 2 2 | _ 3 ++L (83% yield) As this example shows, even secondary halides can be used in this reaction. -

Chapter 20 Enolates / Other Carbanions
Chapter 20 Enolates / other Carbanions C-C bond formation is very important ⇒ larger, more complex organic molecule can be made from smaller ones How? carbon nucleophile + carbon electrophile ⇒ C C C C O δ+ δ- Carbon electrophile : or RC A RX δ+ ⇒ abundant OrgChem- 1 Chap20 How about carbon nucloephile? ⇒ limit ed Ex) cyanide ion: M+ -CN acetylide ion: ylide: Grignard reagent and organo metallic compound: R-X + 2Li → RLi + LiX OrgChem- 2 Chap20 ⇒ Grignard reagent and organometallic compund are not useful for SN2 reactions because they are too reactive. For the SN2 enolates are used Because they are not as reactive as organometallic compunds, then C-C bond through SN2 can be formed OrgChem- 3 Chap20 20.1 Enols and Enolate Anions ⇒ both are possible under acidic and basic conditions In acidic condition In basic condition OrgChem- 4 Chap20 5 Higher value for 1 , 3 -dicarbonyl compounds diketone > ketoester > diester Equilibrium constants of carbonyl and enol tautomers Equilibrium for base condition 20 p OrgChem- Cha What kinds of base can we use? O base O R C CH R C CH R' + BH 2 R' B R''-X O R C CH R' Use base whose pKas of conjugate acids are greater than those of the carbonyl base ! OrgChem- 6 Chap20 For Use compunds whose pKas of conjugate acids are greater than 30 such as For Ethoxide can be used (pKa ~ 16) OrgChem- 7 Chap20 20.2 Halogenation of the α-Carbon The reaction of an aldehyde or ketone with Cl2, Br2, or I2, under either acid or basic condition Both acidic and basis conditions can be used OrgChem- 8 Chap20 Under acidic conditions The presence of the halogens retards the enolation, so the addition of singggple halogen is possible Examples OrgChem- 9 Chap20 Under basic conditions OrgChem- 10 Chap20 Example OrgChem- 11 Chap20 20.3 Alkylation of Enolate Anions ⇒ Enolates from esters, ketones, and nitriles can be used in S N2 reactions (alkylations). -

Conjugate Addition Approach for Natural Product Synthesis Inspired by Gibberellin and Solanoeclepin a Targets*
Pure Appl. Chem., Vol. 85, No. 6, pp. 1149–1160, 2013. http://dx.doi.org/10.1351/PAC-CON-12-10-07 © 2013 IUPAC, Publication date (Web): 18 March 2013 Conjugate addition approach for natural product synthesis inspired by gibberellin and solanoeclepin A targets* Minoru Isobe‡ Department of Chemistry, National Tsing Hua University 101, Section 2, Kuang Fu Road, Hsinchu 30013, Taiwan Abstract: Trimethylzincate is known to react through conjugate addition to α,β-unsaturated ketones, but adds much faster to α,β-unsaturated esters at low temperatures. Since the inter- mediate zinc enolate behaves differently from that of dimethylcuprate, it offers scope for application in a partial synthesis of gibberellin A3. A second example involving vinylsulfones having an oxygen atom on the γ-carbon strongly directs incoming nucleophiles in conjugate addition mode. Heteroatom-directed conjugate addition (HADCA) provides very reactive carbanion intermediates leading to cyclobutane ring formation, necessary for synthesis of solanoeclepin A. An alternative reaction for the four-membered carbocyclic ring closure was explored to make a bond formation between the propargylic cation of Nicholas type and allyltrimethylsilane nucleophile of Hosomi–Sakurai type. This method allowed a formation of tricyclo[5.2.1.01,6]decene framework. Keywords: C–C bond formation; chemical synthesis; cobalt; conjugate addition; gibberellins; organic chemistry; solanoeclepin; stereochemical control; trimethylzincate; vinylsulfone; zinc. INTRODUCTION Conjugate addition has been widely used in organic synthesis, owing to the attendant potential for gen- erating multiple C–C bonds. It may be classified in two categories, the first of which usually entails 1,4-addition to various α,β-unsaturated carbonyl system, and the second of which includes addition to the β-carbon of the olefins conjugating to heteroatom groups. -
Enolate Chemistry I
Dr. P. Wipf Page 1 of 7 9/21/2009 Enolate Chemistry I Cornforth: “Nature is an organic chemist with a preference for the aldol reaction” Many natural products contain polyhydroxylated carbon arrays, usually a mix of 1,2- and 1,3-diols; biosynthetically, these compounds are related and are commonly referred to as polyacetates/polypropionates. Typical examples of polyether antibiotics isolated from Streptomyces are: OMe Me Me Me Me OMe Me Me O Me O Me H O O OMe Me O O O Me Me O O O NH O Me HO N MeO OH Me OH Calcimycin (A-23187) Me Me HO2C NHMe Lonomycin C Me Me Me Et OH Et O OH O H Et O Me H O O Me CO2H Me H Me H Me Me OH OH Lasalocid A OH Me Ionomycin OH O HO Me Me Me H O O HO Me O O O O CH OH MeO H O Me H Et H H H OH 2 Me Me Me Me Me HO C Me 2 Monensin Me Dr. P. Wipf Page 2 of 7 9/21/2009 Most of these compounds are metal chelators (ionophores). Monensin as a food additive kills bacteria in poultry by the transporting Na+ outside the cell, thus increasing the intracellular osmotic pressure and causing the coccidia to explode. Macrolide antibiotics are characterized by the macrocyclic lactone moiety and can be grouped into: polyoxo, polyene, ionophore, and ansamycin macrolides. O O O Me Me Me Me HO HO HO OH Me Me Me Me Me OH Me O Me Me O OR O OR O OR Me O Me Me Me O O OR' Me Me Methymycin Pikromycin Erythromycin A R = desoaminyl R = desoaminyl R = desoaminyl R' = cladinosyl O O Me Me Me CHO CHO MeO Me O OR R'O O OR Me O OH O OH Me Leucomycin A1 Tylosin R = mycarosyl-mycaminosyl R = mycarosyl-mycaminosyl R' = mycinosyl OH OH OH OH O OH O OH HO O OH OH OH OH O HO O OH OH OH OH O CO H CO2H 2 O O OH OH O Amphotericin B O Nystatin A1 NH NH2 2 OH OH Dr. -
Nucleophilic Acyl Substitution
Nucleophilic acyl substitution Nucleophilic acyl substitution describe a class of substitution reactions involving nucleophiles and acyl compounds. In this type of reaction, a nucleophile – such as an alcohol, amine, or enolate – displaces the leaving group of an acyl derivative – such as an acid halide, anhydride, or ester. The resulting product is a carbonyl-containing compound in which the nucleophile has taken the place of the leaving group present in the original acyl derivative. Because acyl derivatives react with a wide variety of nucleophiles, and because the product can depend on the particular type of acyl derivative and nucleophile involved, nucleophilic acyl substitution reactions can be used to synthesize a variety of different products. Contents Reaction mechanism Acidic conditions Basic conditions Reactivity trends Reactions of acyl derivatives Acid halides Thioesters Anhydrides Esters Amides Carboxylic acids See also References External links Reaction mechanism Carbonyl compounds react with nucleophiles via an addition mechanism: the nucleophile attacks the carbonyl carbon, forming a tetrahedral intermediate. This reaction can be accelerated by acidic conditions, which make the carbonyl more electrophilic, or basic conditions, which provide a more anionic and therefore more reactive nucleophile. The tetrahedral intermediate itself can be an alcohol or alkoxide, depending on the pH of the reaction. The tetrahedral intermediate of an acyl compound contains a substituent attached to the central carbon that can act as a leaving group. After the tetrahedral intermediate forms, it collapses, recreating the carbonyl C=O bond and ejecting the leaving group in an elimination reaction. As a result of this two-step addition/elimination process, the nucleophile takes the place of the leaving group on the carbonyl compound by way of an intermediate state that does not contain a carbonyl.