Chap 11. Carbonyl Alpha-Substitution Reactions and Condensation Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry 0310 - Organic Chemistry 1 Chapter 6
Dr. Peter Wipf Chemistry 0310 - Organic Chemistry 1 Chapter 6. SN2 Reactions Nucleophilic substitution reactions occur when nucleophiles (electron-rich species) displace leaving groups on electrophiles (electron-poor species). The net result is the substitution of one group for another bonded to a carbon atom. Nucleophilicity is increased by a negative charge and an increase in the polarizability. The greater the size and the lower the electronegativity of an atom, the greater its polarizability. Leaving groups are stable species that can be detached with their bonding electrons from a molecule during reaction. Most good leaving groups are the conjugate bases of strong acids. Sulfonates (mesylates, tosylates, triflates, etc.) are popular leaving groups since they can be readily obtained from alcohols. Solvents also influence nucleophilicity. SN2 reactions are second-order reactions whose rates depend o the concentration of both the alkyl halide and the nucleophile. The transition state of the one-step SN2 process involves a trigonal bipyramidal carbon and results in an overall inversion of configuration. The order of reactivity of alkyl halides and alkyl sulfonates in SN2 reactions is CH3>1°>2°>3°. The activation energy, DG‡, determines the rate of a reaction: k = koexp(-DG‡/RT). The overall change in free energy, DG, determines the position of the thermodynamic equilibrium. A positive sign of DG is characteristic for an endergonic reaction, a negative DG is found for an exergonic process. The rate of a reaction can be measured and provides information about its mechanism. The reaction order is the sum of the exponents in the rate equation. Relative Leaving Group Abilities: Leaving Group Common Name krel AcO acetate 1 x 10-10 Cl chloride 0.0001 Br bromide 0.001 I iodide 0.01 O mesylate 1.00 H3C S O O O tosylate 0.70 H3C S O O O brosylate 2.62 Br S O O O nosylate 13 O2N S O O O fluorosulfate 29,000 F S O O O triflate 56,000 CF3 S O O O nonaflate 120,000 C4F9 S O O. -

Leaving Group of Substrate Important
1 Recap... • Last two lectures we looked at substitution reactions • We found that there we two (extreme) mechanisms • SN1 H H H H H PhS R H R Br R SPh Br • And SN2 H H H H PhS + Br R Br PhS R • We looked at many of the factors that influenced which ..mechanism was operating... • Now we will turn our attention to elimination reactions 2 Elimination reactions NaOH NaOH or low Br H2O OH concentration • You have seen SN1 substitution • Reaction not sped up by altering nucleophile - it is not in RDS • In fact if we increase the amount / concentration of nucleophile we get the following... high O H + + + Br Br OH concentration H H • We observe an elimination reaction • Overall HBr is lost from the molecule • Isn't chemistry fun - these are the same conditions as substitution! • Next two lectures will look at the mechanisms for elimination • And how we control the nature of the reaction we observe... 3 Elimination, bimolecular E2 O H Br HO Br H H • E2 - Elimination bimolecular or 2nd order reaction • 2 molecules in the RDS or transition state H H H H H C H C H H C Br Br H O H C H O H C C H H H H C H C H H H • Two molecules in the rate determining step • So both the base and the substrate control the reaction • Both carbon skeleton & leaving group of substrate important In substitutions nucleophile acts as a nucleophile & attacks carbon In eliminations nucleophile acts as a base & attacks proton 4 Elimination, bimolecular E2: MO • Little confusing as need bonding & anti-bonding in same diagram! • Orbitals need to be parallel for maximum overlap -

Carbonyl Chemistry I: Additions to Carbonyl Groups
Paul Bracher Chem 30 – Section 4 Carbonyl Chemistry I: Additions to Carbonyl Groups Section Agenda 1) Lessons from Exam I, Course Feedbeck Mechanism on Web Site 2) Carbonyl Groups: Structure, Bonding, Molecular Orbitals, Geometry, Reactivity 3) Handout: In-Section Problem Set 4) Handout: In-Section Solution Set 5) Weekly Office Hours: Thursday, 3-4PM and 8-9PM, in Bauer Lobby Structure and Bonding · Carbonyl carbons are roughly sp2 hybridized Molecular Orbital Picture and planar with bond angles near 120 degrees bonding p orbital antibonding p* orbital · The C=O p bond is formed by the overlap of two unhybridized p orbitals. The bond is polarized due to the different electronegativities of carbon and oxygen. 105 o Resonance Model O O Bürgi-Dunitz angle · Another way to picture carbonyl groups is I II through an MO picture. Since the coefficient of · Resonance form II suggests that the carbon pO’s contribution is greater in pC=O, then atom is surrounded by less negative charge conservation of blending atomic orbitals into density than oxygen, rendering the carbon more molecular orbitals tells you that pC must be a electrophilic. larger contributor to the p*C=O orbital. · When reagents add to carbonyl double bonds, · The larger coefficient on oxygen in the filled the nucleophile attacks the carbon atom. orbital explains why the oxygen atom behaves as a Lewis base (it commonly complexes with · Due to the increased negative charge density Lewis acids). The larger coefficient on carbon on oxygen, Lewis acids (such as protons) in the empty antibonding orbital explains why it commonly complex with oxygen. -
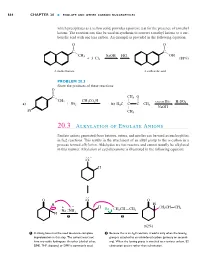
20.3 Alkylation of Enolate Anions
Hornback_Ch20_858-917 12/16/04 12:05 PM Page 864 864 CHAPTER 20 ■ ENOLATE AND OTHER CARBON NUCLEOPHILES which precipitates as a yellow solid, provides a positive test for the presence of a methyl ketone. The reaction can also be used in synthesis to convert a methyl ketone to a car- boxylic acid with one less carbon. An example is provided in the following equation: O O X X C– C– CH3 NaOH HCl OH ϩ 3 Cl2 (88%) A methyl ketone A carboxylic acid PROBLEM 20.3 Show the products of these reactions: O X C– CH3 O CH CH CO H W X 3 ϩ 3 2 – – excess Br2 H2SO4 a) Br2 b) H3C C C CH3 W NaOH Br CH3 20.3 Alkylation of Enolate Anions Enolate anions generated from ketones, esters, and nitriles can be used as nucleophiles in SN2 reactions. This results in the attachment of an alkyl group to the ␣-carbon in a process termed alkylation. Aldehydes are too reactive and cannot usually be alkylated in this manner. Alkylation of cyclohexanone is illustrated in the following equation: . – .O. H . O .O O H – H . œ + – H – œ CH2CH CH2 Na . NH Br CH2CH CH2 H 2 1 2 (62%) 1 A strong base must be used to ensure complete 2 Because this is an SN2 reaction, it works only when the leaving deprotonation in this step. The solvent must not group is attached to an unhindered carbon (primary or second- have any acidic hydrogens. An ether (diethyl ether, ary). When the leaving group is attached to a tertiary carbon, E2 DME, THF, dioxane) or DMF is commonly used. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

Mechanism and Selectivity of Cooperatively Catalyzed Meyer− Schuster Rearrangement/Tsuji−Trost Allylic Substitution
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Article pubs.acs.org/JACS Mechanism and Selectivity of Cooperatively Catalyzed Meyer− Schuster Rearrangement/Tsuji−Trost Allylic Substitution. Evaluation of Synergistic Catalysis by Means of Combined DFT and Kinetics Simulations Marcin Kalek*,† and Fahmi Himo*,‡ † Centre of New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland ‡ Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91 Stockholm, Sweden *S Supporting Information ABSTRACT: The reaction between propargylic alcohols and allylic carbonates, engaging vanadium and palladium catalysts, is an exemplary case of a cooperatively catalyzed process. This combined Meyer−Schuster rearrangement/Tsuji−Trost allylic substitution clearly illustrates the enormous advantages offered by the simultaneous use of two catalysts, but also the inherent challenges regarding selectivity associated with such a reaction design. These challenges originate from the fact that the desired product of the combined process is formed by a bimolecular coupling of the two substrates activated by the respective catalysts. However, these two processes may also occur in a detached way via the reactions of the catalytic intermediates with the starting propargylic alcohol present in the reaction mixture, leading to the formation of two side-products. Herein, we investigate the overall mechanism of this reaction using density functional theory (DFT) methodology. The mechanistic details of the catalytic cycles for all the individual processes are established. In particular, it is shown that the diphosphine ligand, dppm, used in the reaction promotes the formation of dinuclear palladium complexes, wherein only a single metal center is directly involved in the catalysis. -

Allylic Substitution Reactions
Allylic Substitution Reactions! Group Meeting Literature Presentation! Alexanian Research Group! 15 May 2014! ! Njamkou N. Noucti! “Game Changers”! Born in 1927 in Shiga, Japan! Kyoto University (B.S., 1951) ! Columbia University, Gilbert Stork (Ph.D, 1960)! ! Toray Industries, Inc. (1962–1974)! Tokyo Institute of Technology (1974–1988)! Okayama University (1988–1996)! Jiro Tsuji! Kurashiki University of Science and the Arts (1996–1999)! Born in 1941 in Philadelphia, Pennsylvania ! Pennsylvania University (B.S. 1962)! Massachusetts Institute of Technology, Herbert House (Ph.D, 1965)! ! University of Wisconsin–Madison (1965–1987)! Stanford University (1987–Present)! Barry M. Trost! 2! Introduction! “Palladium–catalyzed substitution reactions involving substrates that contain a leaving group in an allylic position”! occur via #–allylmetal intermediates! Pd LG + Nu Nu Historically catalyzed by Palladium but now known with Ir, Mo, W, Ru, and Rh! Allyl fragment in allylic substitution reactions is electrophilic ! NOT TO BE CONFUSED WITH! " Cross–coupling reactions! X MXn + Nucleophilic allyl M = B, Si, Sn, Mg, Zn M = Cl, Br, I, OTs fragments: not allylic " Allylations/Crotylations! substitution O OH reactions! MXn + R R H M = Li, Mg, Sn, Si, B Cr, Ti, Zn, Zr 3! Why Transition–Metal Catalysis?! R R LG + Nu R Nu + SN2 or SN2'! Nu Uncatalyzed allylic substitution reaction! 1) Selectivity (SN2 vs SN2’) is difficult to control without a !catalyst! ! 2)" Transition–metals allow reaction to proceed at lower temperatures! 3)" Catalyzed reactions facilitate asymmetric variants! 4! Outline! !!!!Outline! ! 1." Introduction! 2." History and early developments! 3." Palladium–catalyzed reactions! 4." Iridium–catalyzed reactions! 5." Hard nucleophiles! 6." Conclusion! Topics not covered! Further reading! ! ! Asymmetric Allylic Alkylations! Chem Rev. -

Reactions of Alcohols
Reactions of Alcohols Alcohols are versatile organic compounds since they undergo a wide variety of transformations – the majority of which are either oxidation or reduction type reactions. Normally: Oxidation is a loss of electrons; Reduction is a gain of electrons. But in organic terms: Oxidation: loss of H2; addition of O or O2; addition of X2 (halogens). Reduction: - addition of H2 or H ; loss of O or O2; loss of X2. Neither an oxidation nor reduction: Addition or loss of H+, H2O, HX. Ch11 Reacns of Alcohols (landscape).docx Page 1 Oxidation of Alcohols Primary and secondary alcohols are easily oxidized by a variety of reagents. Secondary Alcohols The most common reagent used for oxidation of secondary alcohols to ketones is chromic acid, H2CrO4. Chromic acid is produced in situ by reaction of sodium dichromate, sulfuric acid and water. Na2Cr2O7 + H2O + 2H2SO4 2 H2CrO4 + 2 NaHSO4 Ch11 Reacns of Alcohols (landscape).docx Page 2 Mechanism of oxidation The alcohol and chromic acid produce a chromate ester, which then reductively eliminates the Cr species. The Cr is reduced (VI IV), the alcohol is oxidized. Oxidation of Primary Alcohols Primary alcohols are easily oxidized just like secondary alcohols, and the INITIAL product of oxidation is an aldehyde. Ch11 Reacns of Alcohols (landscape).docx Page 3 However, the aldehyde can also be easily oxidized to an acid, and this ‘over-oxidation’ is a practical problem. E.g. A common reagent that selectively oxidizes a primary alcohol to an aldehyde (and no further) is pyridinium chlorochromate, PCC. N: CrO3, HCl (PCC) E.g. Tertiary Alcohols These are resistant to oxidation because they have no hydrogen atoms attached to the oxygen bearing carbon (carbinol carbon). -
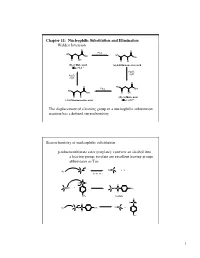
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both.