Reactions of Alcohols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketones Are Simple Organic Compounds Containing a Carbonyl Group
Aldehydes and Ketones are simple organic compounds containing a carbonyl group. Carbonyl group contains carbon- oxygen double bond. These organic compounds are simple because the carbon atom presents in the carbonyl group lack reactive groups such as OH or Cl. By Dr. Sayed Hasan Mehdi Assistant Professor Department of Chemistry Shia P.G. College, Lucknow Dr. S.Hasan Mehdi 6/13/2020 This is to bring to kind notice that the matter of this e- content is for the B.Sc. IV semester students. It has been taken from the following sources. The students are advised to follow these books as well. •A TEXTBOOK OF ORGANIC CHEMISTRY by Arun Bahl & B.S. Bahl, S. Chand & Company Ltd. Publication. •Graduate Organic Chemistry by M. K. Jain and S.C. Sharma, Vishal Publishing Co. •Pradeep’s Organic Chemistry Vol II by R. N. Dhawan, Pradeep Publication, Jalandhar. Dr. S.Hasan Mehdi 6/13/2020 An aldehyde is one of the classes of carbonyl group- containing alkyl group on one end and hydrogen on the other end. The R and Ar denote alkyl or aryl member respectively. In the condensed form, the aldehyde is written as –CHO. Dr. S.Hasan Mehdi 6/13/2020 Dr. S.Hasan Mehdi 6/13/2020 1. From Alcohols: a. By oxidation of Alcohols: Aldehydes and ketones can be prepared by the controlled oxidation of primary and secondary alcohols using an acidified solution of potassium dichromate or permanganate. Primary alcohol produces aldehydesRef. Last slide. O K Cr O RCH2OH + [O] 2 2 7 + R C H H 10 Alcohol Aldehyde O CH3CH2OH+ [O] K2Cr2O7 + CH3 C H H Ethyl Alcohol Acetaldehyde The aldehydes formed in the above reaction are very easily oxidised to carboxylc acids if allowed to remain in the reaction mixture. -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water
Orig Life Evol Biosph (2011) 41:399–412 DOI 10.1007/s11084-011-9243-4 PREBIOTIC CHEMISTRY The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water Paul J. Bracher & Phillip W. Snyder & Brooks R. Bohall & George M. Whitesides Received: 14 April 2011 /Accepted: 16 June 2011 / Published online: 5 July 2011 # Springer Science+Business Media B.V. 2011 Abstract This article reports rate constants for thiol–thioester exchange (kex), and for acid- mediated (ka), base-mediated (kb), and pH-independent (kw) hydrolysis of S-methyl thioacetate and S-phenyl 5-dimethylamino-5-oxo-thiopentanoate—model alkyl and aryl thioalkanoates, respectively—in water. Reactions such as thiol–thioester exchange or aminolysis could have generated molecular complexity on early Earth, but for thioesters to have played important roles in the origin of life, constructive reactions would have needed to compete effectively with hydrolysis under prebiotic conditions. Knowledge of the kinetics of competition between exchange and hydrolysis is also useful in the optimization of systems where exchange is used in applications such as self-assembly or reversible binding. For the alkyl thioester S-methyl thioacetate, which has been synthesized in −5 −1 −1 −1 −1 −1 simulated prebiotic hydrothermal vents, ka = 1.5×10 M s , kb = 1.6×10 M s , and −8 −1 kw = 3.6×10 s . At pH 7 and 23°C, the half-life for hydrolysis is 155 days. The second- order rate constant for thiol–thioester exchange between S-methyl thioacetate and 2- −1 −1 sulfonatoethanethiolate is kex = 1.7 M s . -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides
Proceedings of the Iowa Academy of Science Volume 61 Annual Issue Article 26 1954 Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides B. R. Bluestein Coe College Albert Hybl Coe College Yoshimi Al Nishioka Coe College Let us know how access to this document benefits ouy Copyright ©1954 Iowa Academy of Science, Inc. Follow this and additional works at: https://scholarworks.uni.edu/pias Recommended Citation Bluestein, B. R.; Hybl, Albert; and Nishioka, Yoshimi Al (1954) "Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides," Proceedings of the Iowa Academy of Science, 61(1), 225-232. Available at: https://scholarworks.uni.edu/pias/vol61/iss1/26 This Research is brought to you for free and open access by the Iowa Academy of Science at UNI ScholarWorks. It has been accepted for inclusion in Proceedings of the Iowa Academy of Science by an authorized editor of UNI ScholarWorks. For more information, please contact [email protected]. Bluestein et al.: Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlor Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides By B. R. BLUESTEIN, ALBERT HYBL* AND YosHIMI AL NISHIOKA INTRODUCTION The reaction kinetics of the alcoholysis of substituted benzoyl chlorides was studied. The mechanism of the alcoholysis reaction, which is most generally accepted ( 1), shows that the overall re action should be second-order and that the reaction should be first-order with respect to the acid chloride and first-order with respect to the alcohol. This rate study was carried out using a large excess of alcohol as the solvent, thus obtaining pseudo-first order rate constants, first-order with respect to the acid chloride only. -

Chemistry 0310 - Organic Chemistry 1 Chapter 6
Dr. Peter Wipf Chemistry 0310 - Organic Chemistry 1 Chapter 6. SN2 Reactions Nucleophilic substitution reactions occur when nucleophiles (electron-rich species) displace leaving groups on electrophiles (electron-poor species). The net result is the substitution of one group for another bonded to a carbon atom. Nucleophilicity is increased by a negative charge and an increase in the polarizability. The greater the size and the lower the electronegativity of an atom, the greater its polarizability. Leaving groups are stable species that can be detached with their bonding electrons from a molecule during reaction. Most good leaving groups are the conjugate bases of strong acids. Sulfonates (mesylates, tosylates, triflates, etc.) are popular leaving groups since they can be readily obtained from alcohols. Solvents also influence nucleophilicity. SN2 reactions are second-order reactions whose rates depend o the concentration of both the alkyl halide and the nucleophile. The transition state of the one-step SN2 process involves a trigonal bipyramidal carbon and results in an overall inversion of configuration. The order of reactivity of alkyl halides and alkyl sulfonates in SN2 reactions is CH3>1°>2°>3°. The activation energy, DG‡, determines the rate of a reaction: k = koexp(-DG‡/RT). The overall change in free energy, DG, determines the position of the thermodynamic equilibrium. A positive sign of DG is characteristic for an endergonic reaction, a negative DG is found for an exergonic process. The rate of a reaction can be measured and provides information about its mechanism. The reaction order is the sum of the exponents in the rate equation. Relative Leaving Group Abilities: Leaving Group Common Name krel AcO acetate 1 x 10-10 Cl chloride 0.0001 Br bromide 0.001 I iodide 0.01 O mesylate 1.00 H3C S O O O tosylate 0.70 H3C S O O O brosylate 2.62 Br S O O O nosylate 13 O2N S O O O fluorosulfate 29,000 F S O O O triflate 56,000 CF3 S O O O nonaflate 120,000 C4F9 S O O. -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts
Organic Process Research & Development 2010, 14, 1501–1505 Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts Heiner Eckert* and Johann Auerweck Department of Chemistry, Technische UniVersitaet Muenchen, Lichtenbergstr. 4, Garching 85747, Germany Abstract: Scheme 1. Decomposition of triphosgene (1a) into carbon tetrachloride, carbon dioxide, and 1 equiv of phosgene (3) Phosgene is quantitatively formed from solid triphosgene in a solvent-free and safe process without any reaction heat, catalyzed by planar N-heterocycles with deactivated imino functions. The rate of phosgene generation is adjustable to the rate of phosgene consumption in the subsequent phosgenation reaction by thermal control, catalyst concentration, and in some cases, specific proper- ties of selected metal phthalocyanines. A thermal runaway reaction of this process is impossible. phosgene in most reactions, yet several phosgenation reactions are advantageously carried out with phosgene, that is, when excessive triphosgene is difficult to remove during the reaction Introduction workup because of its high boiling point of over 200 °C. Its Phosgene (3) is a highly useful and versatile chemical in excess can be destroyed by hydrolysis, when phosgenation performing syntheses.1a Although consisting of only four atoms, products are not sensitive to moisture as are carbonates, four important transformations can be carried out with it in carbamates, ureas, diarylketones, alkylhalides, cyanides, and organic -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g. -
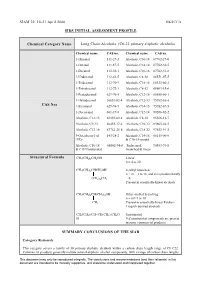
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products.