The Reactions of Alkenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -

Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization Cite this: Chem. Sci.,2018,9, 2898 reactions† Nastaran Salehi Marzijarani,a Roozbeh Yousefi,a Arvind Jaganathan,b Kumar Dilip Ashtekar,a James E. Jackson *a and Babak Borhan *a Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond- forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were + Creative Commons Attribution 3.0 Unported Licence. subjected to this analysis. All showed a strong preference for Cl delivery from the same face of the Received 12th October 2017 alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or Accepted 24th December 2017 carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti DOI: 10.1039/c7sc04430e addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed -

Carboxylic Acids from Limonene Oxidation by Ozone and Hydroxyl Radicals: Insights Into Mechanisms Derived Using a FIGAERO-CIMS
Atmos. Chem. Phys., 19, 13037–13052, 2019 https://doi.org/10.5194/acp-19-13037-2019 © Author(s) 2019. This work is distributed under the Creative Commons Attribution 4.0 License. Carboxylic acids from limonene oxidation by ozone and hydroxyl radicals: insights into mechanisms derived using a FIGAERO-CIMS Julia Hammes1, Anna Lutz1, Thomas Mentel1,2, Cameron Faxon1, and Mattias Hallquist1 1Department of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg, Sweden 2Institute of Energy and Climate Research, Troposphere (IEK-8), Forschungszentrum Jülich GmbH, Jülich, Germany Correspondence: Mattias Hallquist ([email protected]) Received: 21 September 2018 – Discussion started: 27 September 2018 Revised: 10 July 2019 – Accepted: 25 July 2019 – Published: 22 October 2019 Abstract. This work presents the results from a flow reactor are suggested to fill-in the knowledge gaps. Using the addi- study on the formation of carboxylic acids from limonene tional mechanisms proposed in this work, nearly 75 % of the oxidation in the presence of ozone under NOx-free condi- observed gas-phase signal in our lowest concentration exper- tions in the dark. A High-Resolution Time-of-Flight acetate iment (8.4 ppb converted, ca. 23 % acid yield) carried out un- Chemical Ionisation Mass Spectrometer (HR-ToF-CIMS) der humid conditions can be understood. was used in combination with a Filter Inlet for Gases and AEROsols (FIGAERO) to measure the carboxylic acids in the gas and particle phases. The results revealed that limonene oxidation produced large amounts of carboxylic 1 Introduction acids which are important contributors to secondary organic aerosol (SOA) formation. The highest 10 acids contributed Atmospheric aerosol particles have an impact on climate and 56 %–91 % to the total gas-phase signal, and the domi- human health, and their respective effects depend on par- nant gas-phase species in most experiments were C8H12O4, ticle properties determined by the particle size and chem- C9H14O4,C7H10O4 and C10H16O3. -

Development of New Domino Reactions of Alkylidene Meldrum's
Development of New Domino Reactions of Alkylidene Meldrum’s Acids Involving Friedel-Crafts Chemistry and Catalytic Conjugate Allylation of Alkylidene Meldrum’s Acids by Aaron Michael Dumas A thesis presented to the University of Waterloo in fulfilment of the thesis requirement for the degree of Doctor of Philosophy in Chemistry Waterloo, Ontario, Canada, 2009 © Aaron Michael Dumas 2009 I hereby declare that I am the sole author of this thesis. This is a true copy of my thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Alkylidene Meldrum’s acids are very reactive acceptors in conjugate additions, and are known to be significantly more electrophilic than other α,β-unsaturated carbonyl electrophiles. They also offer advantages in terms of ease of preparation, purification and storage. Despite this, they are relatively underused in organic synthesis, and have been treated as something of a curiousity in the literature. The goal of my research was to demonstrate the utility of these molecules in new reactions that are not readily available to other electrophiles. To facilitate this work, new conditions for the Knoevenagel condensation of aldehydes with Meldrum’s acid were developed. This allowed access to a broader range of monosubstituted alkylidenes than was previously possible from any single method. In a reaction that exploits the acylating ability of Meldrum’s acid, a domino addition of phenols to alkylidene Meldrum’s acids was developed. Here, Yb(OTf)3 catalyzed the addition of a phenol to the alkylidene as well as acylation through activation of the electrophile. -

I. an Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of 2011 I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. Charles Edward Schiaffo University of Nebraska-Lincoln Follow this and additional works at: https://digitalcommons.unl.edu/chemistrydiss Part of the Organic Chemistry Commons Schiaffo, Charles Edward, "I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials." (2011). Student Research Projects, Dissertations, and Theses - Chemistry Department. 23. https://digitalcommons.unl.edu/chemistrydiss/23 This Article is brought to you for free and open access by the Chemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Student Research Projects, Dissertations, and Theses - Chemistry Department by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. By Charles E. Schiaffo A DISSERTATION Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Doctor of Philosophy Major: Chemistry Under the Supervision of Professor Patrick H. Dussault Lincoln, Nebraska June, 2011 I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. Charles E. Schiaffo, Ph.D. University of Nebraska-Lincoln, 2011 Advisor: Patrick H. Dussault The use of ozone for the transformation of alkenes to carbonyls has been well established. The reaction of ozone with alkenes in this fashion generates either a 1,2,4- trioxolane (ozonide) or a hydroperoxyacetal, either of which must undergo a separate reduction step to provide the desired carbonyl compound. -

Intensified Ozonolysis of Lignins in a Spray Reactor: Insights Into Product Yields and Lignin Structure
Reaction Chemistry & Engineering Intensified Ozonolysis of Lignins in a Spray Reactor: Insights into Product Yields and Lignin Structure Journal: Reaction Chemistry & Engineering Manuscript ID RE-ART-03-2019-000098.R1 Article Type: Paper Date Submitted by the 20-Apr-2019 Author: Complete List of Authors: Silverman, Julian; University of Kansas, Center for Environmentally Beneficial Catalysis Danby, Andrew; Unviersity of Kansas, Center for Environmentally Beneficial Catalysis Subramaniam, Bala; University of Kansas, Center for Environmentally Beneficial Catalysis Page 1 of 23 Reaction Chemistry & Engineering 1 Intensified Ozonolysis of Lignins in a Spray Reactor: 2 Insights into Product Yields and Lignin Structure 3 Julian R. Silverman1, Andrew M. Danby1, Bala Subramaniam1,2* 4 5 1Center for Environmentally Beneficial Catalysis, University of Kansas, 1501 Wakarusa Drive, 6 Lawrence, Kansas 66047, United States 7 8 2Department of Chemical and Petroleum Engineering, University of Kansas, 1530 W. 15th Street, 9 Lawrence, Kansas 66045, United States 10 11 *Corresponding Author: [email protected]. Tel.: +1-785-864-2903. Fax: +1-785-864-6051 12 13 Abstract 14 We demonstrate a simple spray reactor for an ozonolysis pretreatment step to cleave carbon- 15 carbon double bonds in grass lignins to conveniently recover vanillin and p-hydroxybenzaldehyde 16 (~5 wt. % of the lignin), two of the most value-added monomers. Lignin dissolved in an acid 17 solution is sprayed into an ozone containing gas stream at ambient temperatures with contact 18 times of less than 10 seconds. The production rate of these valuable species is between one to 19 two orders of magnitude greater than that previously reported in a CSTR in which ozone is 20 bubbled through a liquid phase containing dissolved lignin. -

20 More About Oxidation–Reduction Reactions
More About 20 Oxidation–Reduction Reactions OOC n important group of organic reactions consists of those that O A involve the transfer of electrons C from one molecule to another. Organic chemists H OH use these reactions—called oxidation–reduction reactions or redox reactions—to synthesize a large O variety of compounds. Redox reactions are also important C in biological systems because many of these reactions produce HH energy. You have seen a number of oxidation and reduction reactions in other chapters, but discussing them as a group will give you the opportunity to CH3OH compare them. In an oxidation–reduction reaction, one compound loses electrons and one com- pound gains electrons. The compound that loses electrons is oxidized, and the one that gains electrons is reduced. One way to remember the difference between oxidation and reduction is with the phrase “LEO the lion says GER”: Loss of Electrons is Oxi- dation; Gain of Electrons is Reduction. The following is an example of an oxidation–reduction reaction involving inorganic reagents: Cu+ + Fe3+ ¡ Cu2+ + Fe2+ In this reaction,Cu+ loses an electron, so Cu+ is oxidized. Fe3+ gains an electron, so Fe3+ is reduced. The reaction demonstrates two important points about oxidation– reduction reactions. First, oxidation is always coupled with reduction. In other words, a compound cannot gain electrons (be reduced) unless another compound in the reaction simultaneously loses electrons (is oxidized). Second, the compound that is oxidized (Cu+) is called the reducing agent because it loses the electrons that are used to reduce the other compound (Fe3+). Similarly, the compound that is reduced (Fe3+) is called the oxidizing agent because it gains the electrons given up by the other compound (Cu+) when it is oxidized. -

Chapter 8: Reactions of Alkanes; Radicals Page
REACTIONS OF ALKANES: CONTENTS Reactivity Considerations Chlorination and Bromination of Alkanes Reactivity–Selectivity Principle Radical Substitution of Benzylic and Allylic Hydrogens Stereochemistry of Radical Substitution No Radical Reactions in Biological Systems 2 RADICALS Sources include: Hydrogen peroxide . Alkyl peroxides . Light causes homolysis of the weak O-O bond . 3 HETEROLYSIS & HOMOLYSIS Heterolytic bond cleavage or heterolysis H Br H+ + Br Homolytic bond cleavage or homolysis H Br H + Br Homolysis produces radicals, which are very reactive species 4 RADICAL CHAIN REACTIONS Initiation turns stable species into radicals by breaking a bond. 5 RADICAL CHAIN REACTIONS Propagation causes products to form without resulting in net consumption of radicals. (production = consumption) 6 RADICAL CHAIN REACTIONS Termination results in net radical destruction. Generally occurs when reactants are used up. 7 CHLORINATION AND BROMINATION OF ALKANES C H A P T E R 12 8 PRODUCT DISTRIBUTION 9 CHLORINATION RATES 10 CHLORINATION PRODUCT DISTRIBUTION 11 BROMINATION RATES 12 REACTIVITY–SELECTIVITY PRINCIPLE The very reactive chlorine atom will have lower selectivity and attack pretty much any hydrogen available on an alkane The less reactive bromine atom will be more selective and tends to react preferentially with the easy targets, i.e. benzylic/allylic > 3° > 2° > 1° 13 RADICALS Stability of alkyl radicals is similar to stability of carbocations 14 CRUDE RADICAL STABILITY INDEX Add 1 for each attached carbon. Add 3 for adjacent double bond or phenyl ring. Radical equally stable on double bond carbon as on single bonded carbon Profile similar to carbocations but resonance contributes more to radical stability, and radical is OK on C=C. -
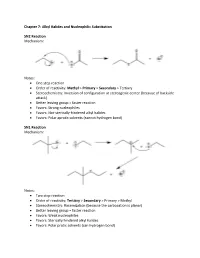
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Download (8MB)
THE MODIFICATION AND USE OF SUPPORTED PLATINUM CATALYSTS FOR ASYMMETRIC HYDROGENATION REACTIONS A Thesis Presented to die University of Glasgow for the Degree of Doctor of Philosophy by Elaine Allan October 1995 ProQuest Number: 11007860 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007860 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 'fCiU l o z U CT 7 G la sg o w i UNIVERSITY ■ i t e y SUMMARY This thesis describes a study of the adsorption of chiral substituted binaphthalene molecules ( 2,2'-dihydroxy-l,l'-binaphthalene, 2,2'-diamino-l,l'-binaphthalene, 2,2- dimethoxy-1,1 '-binaphthalene and 2,2',7,7'-tetrahy droxy-1,1 ’-binaphthalene) on to supported Pt catalysts (1% w/w Pt/y-alumina, 1% w/w Pt/Grace silica CIO and 1% w/w Pt/Cab-O- Sil) with a view to establishing a system which could be capable of inducing asymmetric hydrogenation of prochiral starting materials. The 2,2'-dihydroxy-l,r-binaphthalene modifier was found to adsorb irreversibly on to the 1% w/w Pt/y-alumina and 1% w/w Pt/Grace silica CIO catalysts, prior to the ageing of the 1% w/w Pt/Grace silica CIO catalyst. -

Organic Chemistry I: Reactions and Overview
Organic Chemistry I: Reactions and Overview Andrew Rosen Editor: Raghav Malik January 13, 2013 Contents I Library of Synthetic Reactions 3 II Organic Trends and Essentials 4 1 The Basics: Bonding and Molecular Structure 4 1.1 Resonance Stability . 4 2 Families of Carbon Compounds 4 2.1 Strength of London Dispersion Forces (Polarizability) . 4 2.2 Degree of Unsaturation . 4 3 An Introduction to Organic Reactions and Their Mechanisms 4 3.1 Comparing Acid Strengths . 4 4 Nomenclature and Conformations of Alkanes and Cycloalkanes 5 4.1 Ring Flipping . 5 5 Stereochemistry 5 5.1 Naming Enantiomers via the -R and -S System . 5 5.2 Stereochemistry Examples . 6 6 Ionic Reactions - Overview 6 6.1 General Nucleophilic Substitution Reactions . 6 6.2 Carbocation Stability . 6 6.3 Factors Aecting the Rates of SN 1 and SN 2 Reactions . 6 6.4 Elimination Reactions . 7 6.5 Summary . 7 7 Alkenes and Alkynes I - Overview 8 7.1 The E-Z System . 8 7.2 Relative Stabilities of Alkenes . 8 7.3 Factors Aecting Elimination Reactions . 8 7.4 Acid-Catalyzed Dehydration of Alcohols . 8 1 III Reaction Mechanisms 9 8 Ionic Reactions - Mechanisms 9 8.1 The SN 2 Reaction . 9 8.2 The SN 1 Reaction . 10 8.3 The E2 Reaction . 10 8.4 The E1 Reaction . 11 9 Alkenes and Alkynes I - Mechanisms 11 9.1 Acid-Catalyzed Dehydration of Secondary or Tertiary Alcohols: An E1 Reaction . 11 9.2 Acid-Catalyzed Dehydration of Primary Alcohols: An E2 Reaction . 12 9.3 Synthesis of Alkynes from Vic-Dihalides . -

Reactions of Alcohols
Reactions of Alcohols Alcohols are versatile organic compounds since they undergo a wide variety of transformations – the majority of which are either oxidation or reduction type reactions. Normally: Oxidation is a loss of electrons; Reduction is a gain of electrons. But in organic terms: Oxidation: loss of H2; addition of O or O2; addition of X2 (halogens). Reduction: - addition of H2 or H ; loss of O or O2; loss of X2. Neither an oxidation nor reduction: Addition or loss of H+, H2O, HX. Ch11 Reacns of Alcohols (landscape).docx Page 1 Oxidation of Alcohols Primary and secondary alcohols are easily oxidized by a variety of reagents. Secondary Alcohols The most common reagent used for oxidation of secondary alcohols to ketones is chromic acid, H2CrO4. Chromic acid is produced in situ by reaction of sodium dichromate, sulfuric acid and water. Na2Cr2O7 + H2O + 2H2SO4 2 H2CrO4 + 2 NaHSO4 Ch11 Reacns of Alcohols (landscape).docx Page 2 Mechanism of oxidation The alcohol and chromic acid produce a chromate ester, which then reductively eliminates the Cr species. The Cr is reduced (VI IV), the alcohol is oxidized. Oxidation of Primary Alcohols Primary alcohols are easily oxidized just like secondary alcohols, and the INITIAL product of oxidation is an aldehyde. Ch11 Reacns of Alcohols (landscape).docx Page 3 However, the aldehyde can also be easily oxidized to an acid, and this ‘over-oxidation’ is a practical problem. E.g. A common reagent that selectively oxidizes a primary alcohol to an aldehyde (and no further) is pyridinium chlorochromate, PCC. N: CrO3, HCl (PCC) E.g. Tertiary Alcohols These are resistant to oxidation because they have no hydrogen atoms attached to the oxygen bearing carbon (carbinol carbon).