I. an Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -
A Contribution to the Chemistry of Rhenium
U. S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS RESEARCH PAPER RP999 Part of Journal of Research of the J'Xational Bureau of Standards, Volume 18, May 1937 A CONTRIBUTION TO THE CHEMISTRY OF RHENIUM . By C. E. F. Lundell and H. B. Knowles ABSTRACT A study of the behavior of rhenium when dilute solutions of potassium per rhenate are acidified with sulphuric acid, cooled, and passed through the Jones reductor, indicates that rhenium forms a compound in which it has a valency of minus one, and that the rhenium in this compound is oxidized to a valency of plus one if the diluted sulphuric acid solution is protected from oxygen and warmed to approximately 50° C. In the course of the investigation, it was also found (1) that rhenium can be electrodeposited from diluted (5+95) sulphuric acid solution; (2) that deposits are slightly contaminated; and (3) that the deposited metal can be oxidized directly to perrhenic acid by exposure to moist air, oxygen, or by making the deposit the anode in a water solution. CONTENTS Page 1. Introduction ___ _____________________ ____ __ __ _____ ___ ___ _____ _____ 629 II. ExperimentaL _ ___ _ _ _ _ _ _ _ _ _ _ _ _ __ _ _ __ ___ _ _ _ _ _ __ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ 630 1. Reagents, reductor, and reductor technique ___________________ 630 2. Oxidation of the reduced compound _______________ ___________ 631 3. Potentiometric titrations _______________ ___ ___ ______ ___ _____ 634 4. -

Simulating the Micro Uidic Solvent Extraction of Perrhenate
Simulating the microfluidic solvent extraction of perrhenate Investigating the influence of pH and leakage by Miranda van Duijn to obtain the degree of Bachelor of Science at the Delft University of Technology, to be defended publicly on Tuesday January 23, 2018 at 10:00 AM. Student number: 4355776 Project duration: September, 2017 – January 23, 2018 Thesis committee: Ir. Z. Liu, TU Delft, supervisor Dr. ir. M. Rohde, TU Delft Prof. dr. ir. J.L. Kloosterman TU Delft Abstract Several of the researches conducted at the Reactor Institute Delft (RID) concern the extraction process of perrhenate, ReO4¡. From the perrhenate, rhenium-188 (Re-188) is obtained. Re-188 is medically useful as a high energy ¯ emitter. It can be used as an imaging agent, for in situ tumor treatment and biodistribution. Re-188 is produced via commercially available 188W /188Re radionuclide generators, which have proved their usefulness as a conventional product. Via the production with this generator, the ReO4¡ resides in an aqueous solution together with other compounds such as tungsten-188 (W-188). To medically use the ReO4¡, it needs to be of a certain degree of radionuclidic purity, the proportion of the total radioactivity that is present as a specific radionuclide. [1] A relatively new method to extract the ReO4¡ from the aqueous solution into a liquid organic phase might be microfluidic solvent extraction. This principle is based on the laminar coflowing of two liquid immisci- ble phases between which an interface develops. The ReO4¡ is then transferred through the interface, thus leading to an extraction. Microfluidic solvent extraction offers several inherent advantages, such as the high surface-to-volume ra- tio and short diffusion distances. -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Liquid-Liquid Extraction
OCTOBER 2020 www.processingmagazine.com A BASIC PRIMER ON LIQUID-LIQUID EXTRACTION IMPROVING RELIABILITY IN CHEMICAL PROCESSING WITH PREVENTIVE MAINTENANCE DRIVING PACKAGING SUSTAINABILITY IN THE TIME OF COVID-19 Detecting & Preventing Pressure Gauge AUTOMATIC RECIRCULATION VALVES Failures Schroedahl www.circor.com/schroedahl page 48 LOW-PROFILE, HIGH-CAPACITY SCREENER Kason Corporation www.kason.com page 16 chemical processing A basic primer on liquid-liquid extraction An introduction to LLE and agitated LLE columns | By Don Glatz and Brendan Cross, Koch Modular hemical engineers are often faced with The basics of liquid-liquid extraction the task to design challenging separation While distillation drives the separation of chemicals C processes for product recovery or puri- based upon dif erences in relative volatility, LLE is a fication. This article looks at the basics separation technology that exploits the dif erences in of one powerful and yet overlooked separation tech- the relative solubilities of compounds in two immis- nique: liquid-liquid extraction. h ere are other unit cible liquids. Typically, one liquid is aqueous, and the operations used to separate compounds, such as other liquid is an organic compound. distillation, which is taught extensively in chemical Used in multiple industries including chemical, engineering curriculums. If a separation is feasible by pharmaceutical, petrochemical, biobased chemicals distillation and is economical, there is no reason to and l avor and fragrances, this approach takes careful consider liquid-liquid extraction (LLE). However, dis- process design by experienced chemical engineers and tillation may not be a feasible solution for a number scientists. In many cases, LLE is the best choice as a of reasons, such as: separation technology and well worth searching for a • If it requires a complex process sequence (several quali ed team to assist in its development and design. -

Selection of Thermodynamic Methods
P & I Design Ltd Process Instrumentation Consultancy & Design 2 Reed Street, Gladstone Industrial Estate, Thornaby, TS17 7AF, United Kingdom. Tel. +44 (0) 1642 617444 Fax. +44 (0) 1642 616447 Web Site: www.pidesign.co.uk PROCESS MODELLING SELECTION OF THERMODYNAMIC METHODS by John E. Edwards [email protected] MNL031B 10/08 PAGE 1 OF 38 Process Modelling Selection of Thermodynamic Methods Contents 1.0 Introduction 2.0 Thermodynamic Fundamentals 2.1 Thermodynamic Energies 2.2 Gibbs Phase Rule 2.3 Enthalpy 2.4 Thermodynamics of Real Processes 3.0 System Phases 3.1 Single Phase Gas 3.2 Liquid Phase 3.3 Vapour liquid equilibrium 4.0 Chemical Reactions 4.1 Reaction Chemistry 4.2 Reaction Chemistry Applied 5.0 Summary Appendices I Enthalpy Calculations in CHEMCAD II Thermodynamic Model Synopsis – Vapor Liquid Equilibrium III Thermodynamic Model Selection – Application Tables IV K Model – Henry’s Law Review V Inert Gases and Infinitely Dilute Solutions VI Post Combustion Carbon Capture Thermodynamics VII Thermodynamic Guidance Note VIII Prediction of Physical Properties Figures 1 Ideal Solution Txy Diagram 2 Enthalpy Isobar 3 Thermodynamic Phases 4 van der Waals Equation of State 5 Relative Volatility in VLE Diagram 6 Azeotrope γ Value in VLE Diagram 7 VLE Diagram and Convergence Effects 8 CHEMCAD K and H Values Wizard 9 Thermodynamic Model Decision Tree 10 K Value and Enthalpy Models Selection Basis PAGE 2 OF 38 MNL 031B Issued November 2008, Prepared by J.E.Edwards of P & I Design Ltd, Teesside, UK www.pidesign.co.uk Process Modelling Selection of Thermodynamic Methods References 1. -

Distillation Theory
Chapter 2 Distillation Theory by Ivar J. Halvorsen and Sigurd Skogestad Norwegian University of Science and Technology Department of Chemical Engineering 7491 Trondheim, Norway This is a revised version of an article published in the Encyclopedia of Separation Science by Aca- demic Press Ltd. (2000). The article gives some of the basics of distillation theory and its purpose is to provide basic understanding and some tools for simple hand calculations of distillation col- umns. The methods presented here can be used to obtain simple estimates and to check more rigorous computations. NTNU Dr. ing. Thesis 2001:43 Ivar J. Halvorsen 28 2.1 Introduction Distillation is a very old separation technology for separating liquid mixtures that can be traced back to the chemists in Alexandria in the first century A.D. Today distillation is the most important industrial separation technology. It is particu- larly well suited for high purity separations since any degree of separation can be obtained with a fixed energy consumption by increasing the number of equilib- rium stages. To describe the degree of separation between two components in a column or in a column section, we introduce the separation factor: ()⁄ xL xH S = ------------------------T (2.1) ()x ⁄ x L H B where x denotes mole fraction of a component, subscript L denotes light compo- nent, H heavy component, T denotes the top of the section, and B the bottom. It is relatively straightforward to derive models of distillation columns based on almost any degree of detail, and also to use such models to simulate the behaviour on a computer. -

Cyclotron Produced Radionuclides: Guidelines for Setting up a Facility, Technical Reports Series No
f f f IAEAIAEA RADIOISOTOPESRADIOISOTOPES ANDAND RADIOPHARMACEUTICALSRADIOPHARMACEUTICALS REPORTSREPORTS NNo.. 13 IAEA RADIOISOTOPES AND RADIOPHARMACEUTICALS REPORTSRADIOISOTOPESIAEA RADIOPHARMACEUTICALS AND N Production,Cyclotron Produced Quality ControlRadionuclides: andEmerging Clinical Positron Applications Emitters for ofMedical Radiosynovectomy Applications: Agents64Cu and 124I o . 3 . Atoms for Peace INTERNATIONAL ATOMIC ENERGY AGENCY VIENNA Atoms for Peace Atoms for Peace IAEA RADIOISOTOPES AND Atoms for Peace RADIOPHARMACEUTICALS SERIES PUBLICATIONS One of the main objectives of the IAEA Radioisotope Production and Radiation Technology programme is to enhance the expertise and capability of IAEA Member States in deploying emerging radioisotope products and generators for medical and industrial applications in order to meet national needs as well as to assimilate new developments in radiopharmaceuticals for diagnostic and therapeutic applications. This will ensure local availability of these applications within a framework of quality assurance. Publications in the IAEA Radioisotopes and Radiopharmaceuticals Series provide information in the areas of: reactor and accelerator produced radioisotopes, generators and sealed sources development/production for medical and industrial uses; radiopharmaceutical sciences, including radiochemistry, radiotracer development, production methods and quality assurance/ quality control (QA/QC). The publications have a broad readership and are aimed at meeting the needs of scientists, engineers, -

Distillation Accessscience from McgrawHill Education
6/19/2017 Distillation AccessScience from McGrawHill Education (http://www.accessscience.com/) Distillation Article by: King, C. Judson University of California, Berkeley, California. Last updated: 2014 DOI: https://doi.org/10.1036/10978542.201100 (https://doi.org/10.1036/10978542.201100) Content Hide Simple distillations Fractional distillation Vaporliquid equilibria Distillation pressure Molecular distillation Extractive and azeotropic distillation Enhancing energy efficiency Computational methods Stage efficiency Links to Primary Literature Additional Readings A method for separating homogeneous mixtures based upon equilibration of liquid and vapor phases. Substances that differ in volatility appear in different proportions in vapor and liquid phases at equilibrium with one another. Thus, vaporizing part of a volatile liquid produces vapor and liquid products that differ in composition. This outcome constitutes a separation among the components in the original liquid. Through appropriate configurations of repeated vaporliquid contactings, the degree of separation among components differing in volatility can be increased manyfold. See also: Phase equilibrium (/content/phaseequilibrium/505500) Distillation is by far the most common method of separation in the petroleum, natural gas, and petrochemical industries. Its many applications in other industries include air fractionation, solvent recovery and recycling, separation of light isotopes such as hydrogen and deuterium, and production of alcoholic beverages, flavors, fatty acids, and food oils. Simple distillations The two most elementary forms of distillation are a continuous equilibrium distillation and a simple batch distillation (Fig. 1). http://www.accessscience.com/content/distillation/201100 1/10 6/19/2017 Distillation AccessScience from McGrawHill Education Fig. 1 Simple distillations. (a) Continuous equilibrium distillation. -

Carboxylic Acids from Limonene Oxidation by Ozone and Hydroxyl Radicals: Insights Into Mechanisms Derived Using a FIGAERO-CIMS
Atmos. Chem. Phys., 19, 13037–13052, 2019 https://doi.org/10.5194/acp-19-13037-2019 © Author(s) 2019. This work is distributed under the Creative Commons Attribution 4.0 License. Carboxylic acids from limonene oxidation by ozone and hydroxyl radicals: insights into mechanisms derived using a FIGAERO-CIMS Julia Hammes1, Anna Lutz1, Thomas Mentel1,2, Cameron Faxon1, and Mattias Hallquist1 1Department of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg, Sweden 2Institute of Energy and Climate Research, Troposphere (IEK-8), Forschungszentrum Jülich GmbH, Jülich, Germany Correspondence: Mattias Hallquist ([email protected]) Received: 21 September 2018 – Discussion started: 27 September 2018 Revised: 10 July 2019 – Accepted: 25 July 2019 – Published: 22 October 2019 Abstract. This work presents the results from a flow reactor are suggested to fill-in the knowledge gaps. Using the addi- study on the formation of carboxylic acids from limonene tional mechanisms proposed in this work, nearly 75 % of the oxidation in the presence of ozone under NOx-free condi- observed gas-phase signal in our lowest concentration exper- tions in the dark. A High-Resolution Time-of-Flight acetate iment (8.4 ppb converted, ca. 23 % acid yield) carried out un- Chemical Ionisation Mass Spectrometer (HR-ToF-CIMS) der humid conditions can be understood. was used in combination with a Filter Inlet for Gases and AEROsols (FIGAERO) to measure the carboxylic acids in the gas and particle phases. The results revealed that limonene oxidation produced large amounts of carboxylic 1 Introduction acids which are important contributors to secondary organic aerosol (SOA) formation. The highest 10 acids contributed Atmospheric aerosol particles have an impact on climate and 56 %–91 % to the total gas-phase signal, and the domi- human health, and their respective effects depend on par- nant gas-phase species in most experiments were C8H12O4, ticle properties determined by the particle size and chem- C9H14O4,C7H10O4 and C10H16O3. -

Distillation Design the Mccabe-Thiele Method Distiller Diagam Introduction
Distillation Design The McCabe-Thiele Method Distiller diagam Introduction • UiUsing r igorous tray-bby-tray callilculations is time consuming, and is often unnecessary. • OikhdfiifbOne quick method of estimation for number of plates and feed stage can be obtained from the graphical McCabe-Thiele Method. • This eliminates the need for tedious calculations, and is also the first step to understanding the Fenske-Underwood- Gilliland method for multi-component distillation • Typi ica lly, the in le t flow to the dis tilla tion co lumn is known, as well as mole percentages to feed plate, because these would be specified by plant conditions. • The desired composition of the bottoms and distillate prodillbifiddhiilldducts will be specified, and the engineer will need to design a distillation column to produce these results. • With the McCabe-Thiele Method, the total number of necessary plates, as well as the feed plate location can biddbe estimated, and some ifiinformation can a lblso be determined about the enthalpic condition of the feed and reflux ratio. • This method assumes that the column is operated under constant pressure, and the constant molal overflow assumption is necessary, which states that the flow rates of liquid and vapor do not change throughout the column. • To understand this method, it is necessary to first elaborate on the subjects of the x-y diagram, and the operating lines used to create the McCabe-Thiele diagram. The x-y diagram • The x-y diagram depi icts vapor-liliqu id equilibrium data, where any point on the curve shows the variations of the amount of liquid that is in equilibrium with vapor at different temperatures. -
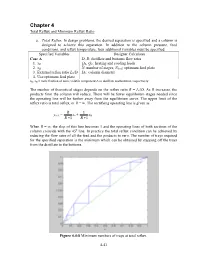
Minimum Reflux Ratio
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux.