20 More About Oxidation–Reduction Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -
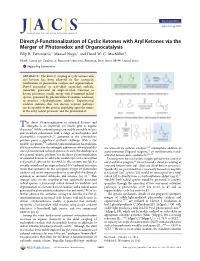
Direct Β‑Functionalization of Cyclic Ketones with Aryl Ketones Via the Merger of Photoredox and Organocatalysis Filip R
Communication pubs.acs.org/JACS Direct β‑Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis Filip R. Petronijevic,́† Manuel Nappi,† and David W. C. MacMillan* Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States *S Supporting Information ABSTRACT: The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl−alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species, generated by photon-induced enamine oxidation, to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst. he direct β-functionalization of saturated ketones and T aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has tradition- ally been restricted to the conjugate addition of soft nucleophiles are accessed via carbene catalysis,9,10 nucleophilic addition of into α,β-unsaturated carbonyl systems. As such, the development acetal-protected Grignard reagents,11 or stoichiometric metal- 5 − of a general catalytic platform for the direct β-functionalization activated homoenolate equivalents.12 16 of saturated ketones or aldehydes would represent a conceptual Drawing from the mechanistic insights gained in the course of and practical advance for the field. -

Process for Purification of Ethylene Compound Having Fluorine-Containing Organic Group
Office europeen des brevets (fi) Publication number : 0 506 374 A1 @ EUROPEAN PATENT APPLICATION @ Application number: 92302586.0 @ Int. CI.5: C07C 17/38, C07C 21/18 (g) Date of filing : 25.03.92 (30) Priority : 26.03.91 JP 86090/91 (72) Inventor : Kishita, Hirofumi 3-19-1, Isobe, Annaka-shi Gunma-ken (JP) (43) Date of publication of application : Inventor : Sato, Shinichi 30.09.92 Bulletin 92/40 3-5-5, Isobe, Annaka-shi Gunma-ken (JP) Inventor : Fujii, Hideki (84) Designated Contracting States : 3-12-37, Isobe, Annaka-shi DE FR GB Gunma-ken (JP) Inventor : Matsuda, Takashi 791 ~4 YdndSG Anri3k3~shi @ Applicant : SHIN-ETSU CHEMICAL CO., LTD. Gunma-ken (JP) 6-1, Ohtemachi 2-chome Chiyoda-ku Tokyo (JP) @) Representative : Votier, Sidney David et al CARPMAELS & RANSFORD 43, Bloomsbury Square London WC1A 2RA (GB) (54) Process for purification of ethylene compound having fluorine-containing organic group. (57) A process for purifying an ethylene compound having a fluorine-containing organic group (fluorine- containing ethylene compound) by mixing the fluorine-containing ethylene compound with an alkali metal or alkaline earth metal reducing agent, and subjecting the resulting mixture to irradiation with ultraviolet radiation, followed by washing with water. The purification process ensures effective removal of iodides which are sources of molecular iodine, from the fluorine-containing ethylene compound. < h- CO CO o 10 o Q_ LU Jouve, 18, rue Saint-Denis, 75001 PARIS EP 0 506 374 A1 BACKGROUND OF THE INVENTION 1. Field of the Invention 5 The present invention relates to a process for purifying an ethylene compound having a fluorine-containing organic group, and more particularly to a purification process by which iodine contained as impurity in an ethylene compound having a fluorine-containing organic group can be removed effectively. -
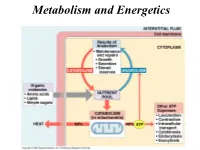
Metabolism and Energetics Oxidation of Carbon Atoms of Glucose Is the Major Source of Energy in Aerobic Metabolism
Metabolism and Energetics Oxidation of carbon atoms of glucose is the major source of energy in aerobic metabolism C6H1206 + 6O2 yields 6 CO2 + H20 + energy Energy released ΔG = - 686 kcal/mol Glucose oxidation requires over 25 discrete steps, with production of 36 ATP. Energy Transformations The mitochondrial synthesis of ATP is not stochiometric. Electron–motive force Proton-motive force Phosphoryl-transfer potential in the form of ATP. Substrate level phosphorylation The formation of ATP by substrate-level phosphorylation ADP ATP P CH O P CH2 O P 2 is used to represent HC OH HC OH a phosphate ester: phosphoglycerate CH OH OH CH2 O P kinase 2 P O bisphosphoglycerate 3-phosphoglycerate OH ADP ATP CH2 CH2 CH3 C O P C OH C O pyruvate kinase non-enzymic COOH COOH COOH phosphoenolpyruvate enolpyruvate pyruvate Why ATP? The reaction of ATP hydrolysis is very favorable ΔGo = - 30.5 kJ/mol = - 7.3 kCal/mol 1. Charge separation of closely packed phosphate groups provides electrostatic relief. Mg2+ 2. Inorganic Pi, the product of the reaction, is immediately resonance-stabilized (electron density spreads equally to all oxygens). 3. ADP immediately ionizes giving H+ into a low [H+] environment (pH~7). 4. Both Pi and ADP are more favorably solvated by water than one ATP molecule. 5. ATP is water soluble. The total body content of ATP and ADP is under 350 mmol – about 10 g, BUT … the amount of ATP synthesized and used each day is about 150 mol – about 110 kg. ATP Production - stage 1 - Glycolysis Glycolysis When rapid production of ATP is needed. -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -

Aldehydes and Ketones Are Simple Organic Compounds Containing a Carbonyl Group
Aldehydes and Ketones are simple organic compounds containing a carbonyl group. Carbonyl group contains carbon- oxygen double bond. These organic compounds are simple because the carbon atom presents in the carbonyl group lack reactive groups such as OH or Cl. By Dr. Sayed Hasan Mehdi Assistant Professor Department of Chemistry Shia P.G. College, Lucknow Dr. S.Hasan Mehdi 6/13/2020 This is to bring to kind notice that the matter of this e- content is for the B.Sc. IV semester students. It has been taken from the following sources. The students are advised to follow these books as well. •A TEXTBOOK OF ORGANIC CHEMISTRY by Arun Bahl & B.S. Bahl, S. Chand & Company Ltd. Publication. •Graduate Organic Chemistry by M. K. Jain and S.C. Sharma, Vishal Publishing Co. •Pradeep’s Organic Chemistry Vol II by R. N. Dhawan, Pradeep Publication, Jalandhar. Dr. S.Hasan Mehdi 6/13/2020 An aldehyde is one of the classes of carbonyl group- containing alkyl group on one end and hydrogen on the other end. The R and Ar denote alkyl or aryl member respectively. In the condensed form, the aldehyde is written as –CHO. Dr. S.Hasan Mehdi 6/13/2020 Dr. S.Hasan Mehdi 6/13/2020 1. From Alcohols: a. By oxidation of Alcohols: Aldehydes and ketones can be prepared by the controlled oxidation of primary and secondary alcohols using an acidified solution of potassium dichromate or permanganate. Primary alcohol produces aldehydesRef. Last slide. O K Cr O RCH2OH + [O] 2 2 7 + R C H H 10 Alcohol Aldehyde O CH3CH2OH+ [O] K2Cr2O7 + CH3 C H H Ethyl Alcohol Acetaldehyde The aldehydes formed in the above reaction are very easily oxidised to carboxylc acids if allowed to remain in the reaction mixture. -

Recent Advances in Titanium Radical Redox Catalysis
JOCSynopsis Cite This: J. Org. Chem. 2019, 84, 14369−14380 pubs.acs.org/joc Recent Advances in Titanium Radical Redox Catalysis Terry McCallum, Xiangyu Wu, and Song Lin* Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States ABSTRACT: New catalytic strategies that leverage single-electron redox events have provided chemists with useful tools for solving synthetic problems. In this context, Ti offers opportunities that are complementary to late transition metals for reaction discovery. Following foundational work on epoxide reductive functionalization, recent methodological advances have significantly expanded the repertoire of Ti radical chemistry. This Synopsis summarizes recent developments in the burgeoning area of Ti radical catalysis with a focus on innovative catalytic strategies such as radical redox-relay and dual catalysis. 1. INTRODUCTION a green chemistry perspective, the abundance and low toxicity of Ti make its complexes highly attractive as reagents and Radical-based chemistry has long been a cornerstone of 5 1 catalysts in organic synthesis. synthetic organic chemistry. The high reactivity of organic IV/III radicals has made possible myriad new reactions that cannot be A classic example of Ti -mediated reactivity is the reductive ring opening of epoxides. This process preferentially readily achieved using two-electron chemistry. However, the − high reactivity of organic radicals is a double-edged sword, as cleaves and functionalizes the more substituted C O bond, the selectivity of these fleeting intermediates can be difficult to providing complementary regioselectivity to Lewis acid control in the presence of multiple chemotypes. In addition, promoted epoxide reactions. The synthetic value of Ti redox catalysis has been highlighted by their many uses in total catalyst-controlled regio- and stereoselective reactions involv- 6−10 ing free-radical intermediates remain limited,2 and the synthesis (Scheme 1). -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Elementary Iodine-Doped Activated Carbon As an Oxidizing Agent for the Treatment of Arsenic-Enriched Drinking Water
water Article Elementary Iodine-Doped Activated Carbon as an Oxidizing Agent for the Treatment of Arsenic-Enriched Drinking Water Fabio Spaziani 1,2,*, Yuli Natori 1, Yoshiaki Kinase 1, Tomohiko Kawakami 1 and Katsuyoshi Tatenuma 1 1 KAKEN Inc., Mito Institute 1044 Hori, Mito, Ibaraki 310-0903, Japan 2 ENEA Casaccia, Via Anguillarese 301, 00123 Roma, Italy * Correspondence: [email protected] Received: 18 July 2019; Accepted: 23 August 2019; Published: 27 August 2019 Abstract: An activated carbon impregnated with elementary iodine (I2), named IodAC, characterized by oxidation capability, was developed and applied to oxidize arsenite, As(III), to arsenate, As(V), in arsenic-rich waters. Batch and column experiments were conducted to test the oxidation ability of the material. Comparisons with the oxidizing agents usually used in arsenic treatment systems were also conducted. In addition, the material has been tested coupled with an iron-based arsenic sorbent, in order to verify its suitability for the dearsenication of drinking waters. IodAC exhibited a high and lasting oxidation potential, since the column tests executed on water spiked with 50 mg/L of arsenic (100% arsenite) showed that 1 cc of IodAC (30 wt% I2) can oxidize about 25 mg of As(III) (0.33 mmol) before showing a dwindling in the oxidation ability. Moreover, an improvement of the arsenic sorption capability of the tested sorbent was also proved. The results confirmed that IodAC is suitable for implementation in water dearsenication plants, in place of the commonly used oxidizing agents, such as sodium hypochlorite or potassium permanganate, and in association with arsenic sorbents. -

Oxidation of Secondary Alcohols to Ketones
Oxidation of secondary alcohols to ketones The oxidation of secondary alcohols to ketones is an important oxidation reaction in organic chemistry. Where a secondary alcohol is oxidised, it is converted to a ketone. The hydrogen from the hydroxyl group is lost along with the hydrogen bonded to the second carbon. The remaining oxygen then forms double bonds with the carbon. This leaves a ketone, as R1–COR2. Ketones cannot normally be oxidised any further because this would involve breaking a C–C bond, which requires too much energy.[1] The reaction can occur using a variety of oxidants. Contents Potassium dichromate PCC (Pyridinium chlorochromate) Dess–Martin oxidation Swern oxidation Oppenauer oxidation Fétizon oxidation See also References Potassium dichromate A secondary alcohol can be oxidised into a ketone using acidified potassium dichromate and heating under 2− 3+ reflux. The orange-red dichromate ion, Cr2O7 , is reduced to the green Cr ion. This reaction was once used in an alcohol breath test. PCC (Pyridinium chlorochromate) PCC, when used in an organic solvent, can be used to oxidise a secondary alcohol into a ketone. It has the advantage of doing so selectively without the tendency to over-oxidise. Dess–Martin oxidation The Dess–Martin periodinane is a mild oxidant for the conversion of alcohols to aldehydes or ketones.[2] The reaction is performed under standard conditions, at room temperature, most often in dichloromethane. The reaction takes between half an hour and two hours to complete. The product is then separated from the spent periodinane.[3] Swern oxidation Swern oxidation oxidises secondary alcohols into ketones using oxalyl chloride and dimethylsulfoxide. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Chapter 14 – Aldehydes and Ketones
Chapter 14 – Aldehydes and Ketones 14.1 Structures and Physical Properties of Aldehydes and Ketones Ketones and aldehydes are related in that they each possess a C=O (carbonyl) group. They differ in that the carbonyl carbon in ketones is bound to two carbon atoms (RCOR’), while that in aldehydes is bound to at least one hydrogen (H2CO and RCHO). Thus aldehydes always place the carbonyl group on a terminal (end) carbon, while the carbonyl group in ketones is always internal. Some common examples include (common name in parentheses): O O H HH methanal (formaldehyde) trans-3-phenyl-2-propenal (cinnamaldehyde) preservative oil of cinnamon O O propanone (acetone) 3-methylcyclopentadecanone (muscone) nail polish remover a component of one type of musk oil Simple aldehydes (e.g. formaldehyde) typically have an unpleasant, irritating odor. Aldehydes adjacent to a string of double bonds (e.g. 3-phenyl-2-propenal) frequently have pleasant odors. Other examples include the primary flavoring agents in oil of bitter almond (Ph- CHO) and vanilla (C6H3(OH)(OCH3)(CHO)). As your book says, simple ketones have distinctive odors (similar to acetone) that are typically not unpleasant in low doses. Like aldehydes, placing a collection of double bonds adjacent to a ketone carbonyl generally makes the substance more fragrant. The primary flavoring agent in oil of caraway is just a such a ketone. 2 O oil of carraway Because the C=O group is polar, small aldehydes and ketones enjoy significant water solubility. They are also quite soluble in typical organic solvents. 14.2 Naming Aldehydes and Ketones Aldehydes The IUPAC names for aldehydes are obtained by using rules similar to those we’ve seen for other functional groups (e.g.