Development of New Domino Reactions of Alkylidene Meldrum's
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization Cite this: Chem. Sci.,2018,9, 2898 reactions† Nastaran Salehi Marzijarani,a Roozbeh Yousefi,a Arvind Jaganathan,b Kumar Dilip Ashtekar,a James E. Jackson *a and Babak Borhan *a Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond- forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were + Creative Commons Attribution 3.0 Unported Licence. subjected to this analysis. All showed a strong preference for Cl delivery from the same face of the Received 12th October 2017 alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or Accepted 24th December 2017 carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti DOI: 10.1039/c7sc04430e addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Regioselective Addition of 1,3-Dicarbonyl Dianion to N-Sulfonyl Aldimines: an Expedient Route to N-Sulfonyl Piperidines and N-Sulfonyl Azetidines
Tetrahedron 63 (2007) 4779–4787 Regioselective addition of 1,3-dicarbonyl dianion to N-sulfonyl aldimines: an expedient route to N-sulfonyl piperidines and N-sulfonyl azetidines Manas K. Ghorai,* Amit Kumar and Sandipan Halder Department of Chemistry, Indian Institute of Technology, Kanpur 208 016, India Received 26 June 2006; revised 23 February 2007; accepted 15 March 2007 Available online 18 March 2007 Abstract—A simple route for the synthesis of d-amino-b-keto esters and d-amino-b-diketones is reported. This involves regioselective addition of 1,3-dianions derived from ethyl acetoacetate and acetyl acetone to N-sulfonyl aldimines. The d-amino-b-keto ester derivatives were further converted into the corresponding N-sulfonyl piperidines and N-sulfonyl azetidines. Ó 2007 Elsevier Ltd. All rights reserved. 1. Introduction In continuation of our research on enolate chemistry and also supported by the related literature on the reactivity of 1,3-di- 1,3-Dicarbonyl dianions have been used extensively in or- carbonyl dianions, it was envisaged that dianions derived ganic synthesis for building ring systems. The anion on the from suitable precursors could be regioselectively added to terminal carbon of the dianion can be regioselectively reacted aldimines to give an easy access to d-amino-b-keto esters18 with 1 equiv of an electrophile resulting in an enolate ion, and their diketone analogues. These compounds could easily which can be trapped by the addition of a second electrophile be converted into various piperidine and azetidine deriva- -

Determination of Solvent Effects on Ketoðenol Equilibria of 1,3-Dicarbonyl Compounds Using NMR: Revisiting a Classic Physical C
In the Laboratory Determination of Solvent Effects on Keto–Enol Equilibria W of 1,3-Dicarbonyl Compounds Using NMR Revisiting a Classic Physical Chemistry Experiment Gilbert Cook* and Paul M. Feltman Department of Chemistry, Valparaiso University, Valparaiso, IN 46383; *[email protected] “The influence of solvents on chemical equilibria was discovered in 1896, simultaneously with the discovery of keto–enol tautomerism in 1,3-dicarbonyl compounds” (1). The solvents were divided into two groups according to their ability to isomerize compounds. The study of the keto–enol tautomerism of β-diketones and β-ketoesters in a variety of solvents using proton NMR has been utilized as a physical Figure 1. The β-dicarbonyl compounds studied in the experiment. chemistry experiment for many years (2, 3). The first reported use of NMR keto–enol equilibria determination was by Reeves (4). This technique has been described in detail in an experiment by Garland, Nibler, and Shoemaker (2). panded (i) to give an in-depth analysis of factors influencing The most commonly used β-diketone for these experi- solvent effects in tautomeric equilibria and (ii) to illustrate ments is acetylacetone (Scheme I). Use of proton NMR is a the use of molecular modeling in determining the origin of viable method for measuring this equilibrium because the a molecule’s polarity. The experiment’s original benefits of tautomeric keto–enol equilibrium is slow on the NMR time using proton NMR as a noninvasive method of evaluating scale, but enol (2a)–enol (2b) tautomerism is fast on this scale equilibrium are maintained. (5). It has been observed that acyclic β-diketones and β- Experimental Procedure ketoesters follow Meyer’s rule of a shift in the tautomeric equi- librium toward the keto tautomer with increasing solvent Observations of the solvent effects for three other 1,3- polarity (6). -

Organic Chemistry II / CHEM 252 Chapter 19 – Synthesis And
Organic Chemistry II / CHEM 252 Chapter 19 – Synthesis and Reactions of β-Dicarbonyl Compounds Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction β-Dicarbonyl compounds have two carbonyl groups separated by a carbon • Protons on the α-carbon of β-dicarbonyl compounds are acidic (pKa = 9-10) – The acidity can be explained by resonance stabilization of the corresponding enolate by two carbonyl groups 2 Synthesis Claisen condensation • The acetoacetic ester and malonic acid syntheses use β-dicarbonyl compounds for carbon-carbon bond forming reactions • The acetoacetic ester and malonic ester syntheses usually conclude with decarboxylation of a β-keto acid 3 Synthesis • The Claisen Condensation: Synthesis of β-Keto Esters • Ethyl acetate undergoes a Claisen condensation when treated with sodium ethoxide – The product is commonly called an acetoacetic ester • Ethyl pentanoate undergoes an analogous reaction 4 Synthesis • The overall reaction involves loss of an α hydrogen from one ester and loss of ethoxide from another • The mechanism is an example of the general process of nucleophilic addition-elimination at an ester carbonyl 5 Synthesis 6 Synthesis • The alkoxide base must have the same alkyl group as the alkoxyl group of the ester – The use of a different alkoxide would result in formation of some transesterification products • Esters with only one α hydrogen do not undergo Claisen condensation – A second hydrogen on the α carbon is necessary so that it can be deprotonated in Step 3 – This deprotonation -

Chapter 8: Reactions of Alkanes; Radicals Page
REACTIONS OF ALKANES: CONTENTS Reactivity Considerations Chlorination and Bromination of Alkanes Reactivity–Selectivity Principle Radical Substitution of Benzylic and Allylic Hydrogens Stereochemistry of Radical Substitution No Radical Reactions in Biological Systems 2 RADICALS Sources include: Hydrogen peroxide . Alkyl peroxides . Light causes homolysis of the weak O-O bond . 3 HETEROLYSIS & HOMOLYSIS Heterolytic bond cleavage or heterolysis H Br H+ + Br Homolytic bond cleavage or homolysis H Br H + Br Homolysis produces radicals, which are very reactive species 4 RADICAL CHAIN REACTIONS Initiation turns stable species into radicals by breaking a bond. 5 RADICAL CHAIN REACTIONS Propagation causes products to form without resulting in net consumption of radicals. (production = consumption) 6 RADICAL CHAIN REACTIONS Termination results in net radical destruction. Generally occurs when reactants are used up. 7 CHLORINATION AND BROMINATION OF ALKANES C H A P T E R 12 8 PRODUCT DISTRIBUTION 9 CHLORINATION RATES 10 CHLORINATION PRODUCT DISTRIBUTION 11 BROMINATION RATES 12 REACTIVITY–SELECTIVITY PRINCIPLE The very reactive chlorine atom will have lower selectivity and attack pretty much any hydrogen available on an alkane The less reactive bromine atom will be more selective and tends to react preferentially with the easy targets, i.e. benzylic/allylic > 3° > 2° > 1° 13 RADICALS Stability of alkyl radicals is similar to stability of carbocations 14 CRUDE RADICAL STABILITY INDEX Add 1 for each attached carbon. Add 3 for adjacent double bond or phenyl ring. Radical equally stable on double bond carbon as on single bonded carbon Profile similar to carbocations but resonance contributes more to radical stability, and radical is OK on C=C. -
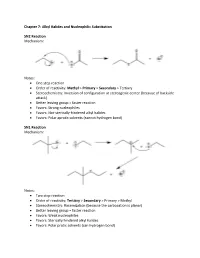
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Download (8MB)
THE MODIFICATION AND USE OF SUPPORTED PLATINUM CATALYSTS FOR ASYMMETRIC HYDROGENATION REACTIONS A Thesis Presented to die University of Glasgow for the Degree of Doctor of Philosophy by Elaine Allan October 1995 ProQuest Number: 11007860 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007860 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 'fCiU l o z U CT 7 G la sg o w i UNIVERSITY ■ i t e y SUMMARY This thesis describes a study of the adsorption of chiral substituted binaphthalene molecules ( 2,2'-dihydroxy-l,l'-binaphthalene, 2,2'-diamino-l,l'-binaphthalene, 2,2- dimethoxy-1,1 '-binaphthalene and 2,2',7,7'-tetrahy droxy-1,1 ’-binaphthalene) on to supported Pt catalysts (1% w/w Pt/y-alumina, 1% w/w Pt/Grace silica CIO and 1% w/w Pt/Cab-O- Sil) with a view to establishing a system which could be capable of inducing asymmetric hydrogenation of prochiral starting materials. The 2,2'-dihydroxy-l,r-binaphthalene modifier was found to adsorb irreversibly on to the 1% w/w Pt/y-alumina and 1% w/w Pt/Grace silica CIO catalysts, prior to the ageing of the 1% w/w Pt/Grace silica CIO catalyst. -

A Thermodynamic Atlas of Carbon Redox Chemical Space
A thermodynamic atlas of carbon redox chemical space Adrian Jinicha,b,1, Benjamin Sanchez-Lengelinga, Haniu Rena, Joshua E. Goldfordc, Elad Noord, Jacob N. Sanderse, Daniel Segrèc,f,g,h, and Alán Aspuru-Guziki,j,k,l,1 aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138; bDivision of Infectious Diseases, Weill Department of Medicine, Weill-Cornell Medical College, New York, NY 10021; cBioinformatics Program and Biological Design Center, Boston University, Boston, MA 02215; dInstitute of Molecular Systems Biology, ETH Zürich, 8093 Zürich, Switzerland; eDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095; fDepartment of Biology, Boston University, Boston, MA 02215; gDepartment of Biomedical Engineering, Boston University, Boston, MA 02215; hDepartment of Physics, Boston University, Boston, MA 02215; iChemical Physics Theory Group, Department of Chemistry, University of Toronto, Toronto, ON, M5S 3H6 Canada; jDepartment of Computer Science, University of Toronto, Toronto, ON, M5T 3A1 Canada; kCIFAR AI Chair, Vector Institute, Toronto, ON, M5S 1M1 Canada; and lLebovic Fellow, Canadian Institute for Advanced Research, Toronto, ON M5S 1M1 Canada Edited by Pablo G. Debenedetti, Princeton University, Princeton, NJ, and approved November 17, 2020 (received for review April 9, 2020) Redox biochemistry plays a key role in the transduction of central metabolic redox substrates (3). In addition, since its chemical energy in living systems. However, the compounds standard potential is ∼100 mV lower than that of the typical observed in metabolic redox reactions are a minuscule fraction aldehyde/ketone functional group, it effectively decreases the of chemical space. It is not clear whether compounds that ended steady-state concentration of potentially damaging aldehydes/ up being selected as metabolites display specific properties that ketones in the cell (3). -

Biosynthesis of Acetylacetone Inspired by Its Biodegradation
Zhou et al. Biotechnol Biofuels (2020) 13:88 https://doi.org/10.1186/s13068-020-01725-9 Biotechnology for Biofuels RESEARCH Open Access Biosynthesis of acetylacetone inspired by its biodegradation Yifei Zhou1,2, Yamei Ding3, Wenjie Gao1, Jichao Wang1, Xiutao Liu1,2, Mo Xian1, Xinjun Feng1* and Guang Zhao1,4* Abstract Background: Acetylacetone is a commercially bulk chemical with diverse applications. However, the traditional manufacturing methods sufer from many drawbacks such as multiple steps, harsh conditions, low yield, and environ- mental problems, which hamper further applications of petrochemical-based acetylacetone. Compared to conven- tional chemical methods, biosynthetic methods possess advantages such as being eco-friendly, and having mild conditions, high selectivity and low potential costs. It is urgent to develop biosynthetic route for acetylacetone to avoid the present problems. Results: The biosynthetic pathway of acetylacetone was constructed by reversing its biodegradation route, and the acetylacetone was successfully produced by engineered Escherichia coli (E. coli) by overexpression of acetylac- etone-cleaving enzyme (Dke1) from Acinetobacter johnsonii. Several promising amino acid residues were selected for enzyme improvement based on sequence alignment and structure analysis, and the acetylacetone production was improved by site-directed mutagenesis of Dke1. The double-mutant (K15Q/A60D) strain presented the highest acetylacetone-producing capacity which is 3.6-fold higher than that of the wild-type protein. Finally, the strain accu- mulated 556.3 15.2 mg/L acetylacetone in fed-batch fermentation under anaerobic conditions. ± Conclusions: This study presents the frst intuitive biosynthetic pathway for acetylacetone inspired by its biodegra- dation, and shows the potential for large-scale production. -

View a Complete List of Ph.D Degrees
1913 1. CLARK, Clinton Willard. (Title not Available). 2. CLARK, Hugh. An Improved Method for the Manufacture of Hydrogen and Lamp Black. 3. CORLISS, Harry Percival. The Distribution of Colloidal Arsenic Trisulfide between the Phases in the System Ether, Water, and Alcohol. 4. PRATT, Lester Albert. Studies in the Field of Petroleum. 5. SHIVELY, Robert Rex. A Study of Magnesia Cements. 1914 6. UHLINGER, Roy H. The Formation of Utilizable Products from Natural Gas. 1915 7. DUNPHY, Raymond Augustine. Theory of Distillation and the Laws of Henry and Raoult. 8. LIEBOVITZ, Sidney. Study of Dynamics of Esterification. 9. MORTON, Harold Arthur. Study of the Specific Rotatory Power of Organic Substances in Solution. 10. WITT, Joshua Chitwood. Oxidation and Reduction Without the Addition of Acid. I. The Reaction between Ferrous Sulfate and Potassium Dichromate. II. The Reaction Between Stanous Chloride and Potassium Dichromate. (Neidle) 1917 11. COLEMAN, Arthur Bert. Tetraiodofluorescein, Tetraiodoeosin, Tetraiodoerythrosin and Some of Their Derivatives. (Pratt) 12. MILLER, Rolla Woods. On the Mechanism of the Potassium Chlorate-Manganese Dioxide Reaction. 13. PERKINS, Granville Akers. Phthalic Anhydride and Some of Its Derivatives. (Pratt) 14. SHUPP, Asher Franklin. Phenoltetraiodophthalein and Some of Its Derivatives. (Pratt) 1919 15. CURME, Henry R. Butadiine (Diacetylene). II. Analysis of Gas Mixtures by Distillation at Low Temperatures and Low Pressures. III. The Precise Analytical Determination of Acetylene, Ethylene, and Methyl Acetylene in Hydrocarbon Gas Mixtures. 16. DROGIN, Isaac. The Effect of Potassium Chloride on the Inversion of Cane Sugar by Formic Acid. 17. YOUNG, Charles Otis. Tetrabromophthalic Acid and Its Condensation with Some Primary Amines. 1 1920 18. -

I the Tandem Chain Extension-Acylation Reaction II Synthesis of Papyracillic Acid A
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Fall 2013 I The tandem chain extension-acylation reaction II Synthesis of papyracillic acid A: Application of the tandem homologation- acylation reaction III Synthesis of tetrahydrofuran-based peptidomimetics Carley Meredith Spencer Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Spencer, Carley Meredith, "I The tandem chain extension-acylation reaction II Synthesis of papyracillic acid A: Application of the tandem homologation-acylation reaction III Synthesis of tetrahydrofuran-based peptidomimetics" (2013). Doctoral Dissertations. 749. https://scholars.unh.edu/dissertation/749 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. I. THE TANDEM CHAIN EXTEN SION - AC YL ATION REACTION II. SYNTHESIS OF PAPYRACILLIC ACID A: APPLICATION OF THE TANDEM HOMOLOGATION-ACYLATION REACTION III. SYNTHESIS OF TETRAHYDROFURAN-BASED PEPTIDOMIMETICS BY Carley Meredith Spencer B.A., Connecticut College, 2008 DISSERTATION Submitted to the University of New Hampshire in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry September 2013 UMI Number: 3575989 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion.