Transcription 12.01.12
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

Enhanced Butanol Production by Free and Immobilized Clostridium Sp
Enhanced Butanol Production by Free and Immobilized Clostridium sp. Cells Using Butyric Acid as Co-Substrate Laili Gholizadeh This thesis comprises 30 ECTS credits and is a compulsory part in the Master of Science with a Major in Chemical Engineering – Applied Biotechnology 120 ECTS credits No. 10/2009 Title: Enhanced Butanol Production by Free and Immobilized Clostridium sp. Cells using Butyric Acid as Co-Substrate. Author: Laili Gholizadeh Baroghi (e-mail: [email protected]) Master Thesis Subject Category: Biotechnology (Bioprocess Engineering – Biofuels) University College of Borås School of Engineering SE-501 90 BORÅS Telephone: (+46) 033 435 4640 Examiner: Prof. Mohammad Taherzadeh Supervisor and Thesis Advisor: Prof. Shang–Tian Yang Supervisor Address: OSU–Ohio State University 125 Koffolt Laboratories 140 West 19th Ave. Columbus, OH 43210–1185, USA Client: Ohio State University (OSU), Chemical & Biomolecular Engineering Department Prof. Shang–Tian Yang Columbus, Ohio; USA. Date: 08–12–2009 Keywords: Bio-butanol y Acetone–Butanol–Ethanol (ABE) y ABE- fermentation y Butyric acid y Clostridium y C. acetobutylicum ATCC 824 y C. beijerinckii ATCC 55025 y C. beijerinckii BA 101 y C. beijerinckii NCIMB 8052 y Fibrous-bed Bioreactor (FBB) y Batch y Suspended cell culture y Immobilized cell system. DEDICATION I would like to dedicate this M.Sc. Thesis to my beloved Family for all their love and encouragement and for always been supportive of my choices. “I am among those who think that science has great beauty. A scientist in his laboratory is not only a technician: he is also a child placed before natural phenomena, which impress him like a fairy tale.” − Marie Curie ABSTRACT Butanol production by four different Clostridium sp. -
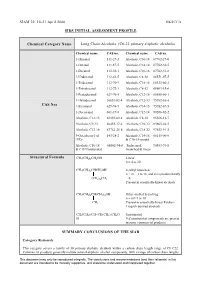
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Aldehydes and Ketones
Organic Lecture Series Aldehydes And Ketones Chap 16 111 Organic Lecture Series IUPAC names • the parent alkane is the longest chain that contains the carbonyl group • for ketones, change the suffix -e to -one • number the chain to give C=O the smaller number • the IUPAC retains the common names acetone, acetophenone, and benzophenone O O O O Propanone Acetophenone Benzophenone 1-Phenyl-1-pentanone (Acetone) Commit to memory 222 Organic Lecture Series Common Names – for an aldehyde , the common name is derived from the common name of the corresponding carboxylic acid O O O O HCH HCOH CH3 CH CH3 COH Formaldehyde Formic acid Acetaldehyde Acetic acid – for a ketone , name the two alkyl or aryl groups bonded to the carbonyl carbon and add the word ketone O O O Ethyl isopropyl ketone Diethyl ketone Dicyclohexyl ketone 333 Organic Lecture Series Drawing Mechanisms • Use double-barbed arrows to indicate the flow of pairs of e - • Draw the arrow from higher e - density to lower e - density i.e. from the nucleophile to the electrophile • Removing e - density from an atom will create a formal + charge • Adding e - density to an atom will create a formal - charge • Proton transfer is fast (kinetics) and usually reversible 444 Organic Lecture Series Reaction Themes One of the most common reaction themes of a carbonyl group is addition of a nucleophile to form a tetrahedral carbonyl addition compound (intermediate). - R O Nu - + C O Nu C R R R Tetrahedral carbonyl addition compound 555 Reaction Themes Organic Lecture Series A second common theme is -

N-BUTYL ALCOHOL
Right to Know Hazardous Substance Fact Sheet Common Name: n-BUTYL ALCOHOL Synonyms: Propyl Carbinol; n-Butanol CAS Number: 71-36-3 Chemical Name: 1-Butanol RTK Substance Number: 1330 Date: November 1998 Revision: January 2008 DOT Number: UN 1120 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE n-Butyl Alcohol is a colorless liquid with a strong, sweet Hazard Summary alcohol odor. It is used as a solvent for fats, waxes, shellacs, Hazard Rating NJDOH NFPA resins, gums, and varnish, in making hydraulic fluids, and in HEALTH - 2 medications for animals. FLAMMABILITY - 3 REACTIVITY - 0 f ODOR THRESHOLD = 1 to 15 ppm FLAMMABLE f Odor thresholds vary greatly. Do not rely on odor alone to POISONOUS GASES ARE PRODUCED IN FIRE determine potentially hazardous exposures. CONTAINERS MAY EXPLODE IN FIRE Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; Reasons for Citation 4=severe f n-Butyl Alcohol is on the Right to Know Hazardous f n-Butyl Alcohol can affect you when inhaled and by Substance List because it is cited by OSHA, ACGIH, DOT, passing through the skin. NIOSH, DEP, IRIS, NFPA and EPA. f Contact can irritate and burn the skin. f This chemical is on the Special Health Hazard Substance f n-Butyl Alcohol can irritate and burn the eyes with possible List. eye damage. f Inhaling n-Butyl Alcohol can irritate the nose, throat and lungs. f Exposure to n-Butyl Alcohol can cause headache, dizziness, nausea and vomiting. SEE GLOSSARY ON PAGE 5. f n-Butyl Alcohol can damage the liver, kidneys, hearing, and sense of balance. -

Reactions of Alcohols & Ethers 1
REACTIONS OF ALCOHOLS & ETHERS 1. Combustion (Extreme Oxidation) alcohol + oxygen carbon dioxide + water 2 CH3CH2OH + 6 O2 4 CO2 + 6 H2O 2. Elimination (Dehydration) ° alcohol H 2 S O 4/ 1 0 0 C alkene + water H2SO4/100 ° C CH3CH2CH2OH CH3CH=CH2 + H2O 3. Condensation ° excess alcohol H 2 S O 4 / 1 4 0 C ether + water H2SO4/140 ° C 2 CH3CH2OH CH3CH2OCH2CH3 + H2O 4. Substitution Lucas Reagent alcohol + hydrogen halide Z n C l2 alkyl halide + water ZnCl2 CH3CH2OH + HCl CH3CH2Cl + H2O • This reaction with the Lucas Reagent (ZnCl2) is a qualitative test for the different types of alcohols because the rate of the reaction differs greatly for a primary, secondary and tertiary alcohol. • The difference in rates is due to the solubility of the resulting alkyl halides • Tertiary Alcohol→ turns cloudy immediately (the alkyl halide is not soluble in water and precipitates out) • Secondary Alcohol → turns cloudy after 5 minutes • Primary Alcohol → takes much longer than 5 minutes to turn cloudy 5. Oxidation • Uses an oxidizing agent such as potassium permanganate (KMnO4) or potassium dichromate (K2Cr2O7). • This reaction can also be used as a qualitative test for the different types of alcohols because there is a distinct colour change. dichromate → chromium 3+ (orange) → (green) permanganate → manganese (IV) oxide (purple) → (brown) Tertiary Alcohol not oxidized under normal conditions CH3 KMnO4 H3C C OH NO REACTION K2Cr2O7 CH3 tertbutyl alcohol Secondary Alcohol ketone + hydrogen ions H O KMnO4 H3C C CH3 + K2Cr2O7 C + 2 H H3C CH3 OH propanone 2-propanol Primary Alcohol aldehyde + water carboxylic acid + hydrogen ions O KMnO4 O H H KMnO H H 4 + CH3CH2CH2OH C C C H C C C H + 2 H K2Cr2O7 + H2O K Cr O H H H 2 2 7 HO H H 1-propanol propanal propanoic acid 6. -

Alcoholsalcohols
AlcoholsAlcohols Chapter 10 1 Structure of Alcohols • The functional group of an alcohol is H an -OH group bonded to an sp3 O 108.9° hybridized carbon. C H – Bond angles about the hydroxyl oxygen H H atom are approximately 109.5°. • Oxygen is sp3 hybridized. –Two sp3 hybrid orbitals form sigma bonds to a carbon and a hydrogen. – The remaining two sp3 hybrid orbitals each contain an unshared pair of electrons. 2 Nomenclature of Alcohols • IUPAC names – The parent chain is the longest chain that contains the OH group. – Number the parent chain to give the OH group the lowest possible number. – Change the suffix -etoe -ol.ol • Common names – Name the alkyl group bonded to oxygen followed by the word alcohol.alcohol 3 Nomenclature of Alcohols OH o o 1o OH 1 2 OH 1-Propanol 2-Propan ol 1-Bu tanol (Pro py l alco ho l) (Isoprop yl alcoh ol) (Bu tyl alcoh ol) OH 2o OH 1o OH 3o 2-Butanol 2-M eth yl-1-p ropan ol 2-M eth yl-2-p ropan ol (sec-Butyl alcohol) (Isobutyl alcohol) (tert -Butyl alcohol) 4 Nomenclature of Alcohols • Compounds containing more than one OH group are named diols, triols, etc. CH2 CH2 CH3 CHCH2 CH2 CHCH2 OH OH HO OH HO HO OH 1,2-Ethanediol 1,2-Propanediol 1,2,3-Propanetriol (Ethylene glycol) (Propylene glycol) (Glycerol, Glycerine) 5 Physical Properties • Alcohols are polar compounds. – They interact with themselves and with other polar compounds by dipole-dipole interactions. • Dipole-dipole interaction: The attraction between the positive end of one dipole and the negative end of another. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Soln-2212-PT3-S20-Ch-16-18.Pdf
1. 2. H2SO4 H2SO4 H2SO41) BH3, THF 1) BH3, THF 1) BH3, THF - - - 2) OH , H2O2, H2O2) OH , H2O2, H2O 2) OH , H2O2, H2O OH OH OH 3. OH OH OH H2SO4 1) BH3, THF - 2) OH , H2O2, H2O OH PBr3 PBr3 PBr3 Pyridine Pyridine Pyridine OH PBr3 Pyridine O O O Mg Mg Mg HO HO HO1) 1) 1) Ether Ether Ether 2) HCl 2) HCl 2) HCl Br Br Br MgBr MgBr MgBr O Mg HO 1) Ether 2) HCl Br 4. MgBr **This reaction reduces an aldehyde to a primary alcohol and a ketone to a secondary alcohol. Since only one EQ of reagent is given the most reactive functional group will be reduced (in this case the aldehyde) **ketones are less reactive than aldehydes and the more steric hindrance surrounding the ketone the less reactive it is. Note: NaBH4 reduces ketones but is too weak to reduce an ester/double/triple bonds 5. ** With these reagents a tertiary amide will be reduced to a tertiary amine, a secondary amide will be reduced to a secondary amine, and a primary amide will be reduced to a primary amine 6. ** Recall from chapter 7, OChem 1 H2 and Lindlars catalyst always reduces an alkyne to a CIS ALKENE ** Li/Na and liquid NH3 always reduces an alkyne to a TRANS ALKENE 7. **A ketone/aldehyde reacts with primary amines in trace acid to produce an imine, and reacts with a secondary amine in trace acid to produce an enamine (double bond forms between the alpha and beta carbons) **Sodium cyanoborohydride (NaBH3CN) is used to reduce imines/enamines back to their respective secondary/tertiary amines 8. -

Strategies to Introduce N-Butanol in Gasoline Blends
sustainability Article Strategies to Introduce n-Butanol in Gasoline Blends Magín Lapuerta *, Rosario Ballesteros and Javier Barba Escuela Técnica Superior de Ingenieros Industriales, University of Castilla-La Mancha, Av. Camilo José Cela s/n, 13071 Ciudad Real, Spain; [email protected] (R.B.); [email protected] (J.B.) * Correspondence: [email protected] Academic Editors: Gilles Lefebvre and Francisco J. Sáez-Martínez Received: 13 February 2017; Accepted: 7 April 2017; Published: 12 April 2017 Abstract: The use of oxygenated fuels in spark ignition engines (SIEs) has gained increasing attention in the last few years, especially when coming from renewable sources, due to the shortage of fossil fuels and global warming concern. Currently, the main substitute of gasoline is ethanol, which helps to reduce CO and HC emissions but presents a series of drawbacks such as a low heating value and a high hygroscopic tendency, which cause higher fuel consumption and corrosion problems, respectively. This paper shows the most relevant properties when replacing ethanol by renewable n-butanol, which presents a higher heating value and a lower hygroscopic tendency compared to the former. The test matrix carried out for this experimental study includes, on the one hand, ethanol substitution by n-butanol in commercial blends and, on the other hand, either ethanol or gasoline substitution by n-butanol in E85 blends (85% ethanol-15% gasoline by volume). The results show that the substitution of n-butanol by ethanol presents a series of benefits such as a higher heating value and a greater interchangeability with gasoline compared to ethanol, which makes n-butanol a promising fuel for SIEs in commercial blends. -

Alcohols I 1400
ALCOHOLS I 1400 Table 1 MW: Table 1 CAS: Table 2 RTECS: Table 2 METHOD: 1400, Issue 2 EVALUATION: PARTIAL Issue 1: 15 February 1984 Issue 2: 15 August 1994 OSHA : Table 2 PROPERTIES: Table 1 NIOSH: Table 2 ACGIH: Table 2 COMPOUNDS AND SYNONYMS: (1) ethanol: ethyl alcohol. (2) isopropyl alcohol: 2-propanol. (3) tert-butyl alcohol: 2-methyl-2-propanol. SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPHY, FID (coconut shell charcoal, 100 mg/50 mg) ANALYTE: compounds above FLOW RATE: 0.01 to 0.2 L/min (£0.05 L/min for ethyl alcohol) DESORPTION: 1 mL 1% 2-butanol in CS 2 (1) (2) (3) INJECTION VOL-MIN: 0.1 L 0.3 L 1.0 L VOLUME: 5 µL -MAX: 1 L 3 L 10 L TEMPERATURE-INJECTION: 200 °C SHIPMENT: cooled -DETECTOR: 250-300 °C -COLUMN: 65-70 °C SAMPLE STABILITY: unknown, store in freezer CARRIER GAS: N2 or He, 30 mL/min BLANKS: 2 to 10 field blanks per set COLUMN: glass, 2 m x 4-mm ID, 0.2% Carbowax 1500 on 60/80 Carbopack C or equivalent ACCURACY CALIBRATION: solutions of analyte in eluent (internal standard optional) RANGE STUDIED: see EVALUATION OF METHOD RANGE AND BIAS: not significant [1] PRECISION: see EVALUATION OF METHOD OVERALL PRECISION (S ˆ ): see EVALUATION OF METHOD rT ESTIMATED LOD: 0.01 mg per sample [2] ACCURACY: ± 14% APPLICABILITY: The working ranges are 16 to 1000 ppm ethanol (30 to 1900 mg/m 3) for a 1-L air sample; 4 to 400 ppm isopropyl alcohol (10 to 1000 mg/m 3) for a 3-L air sample; and 1 to 100 ppm t-butyl alcohol (3 to 300 mg/m 3) for a 10-L air sample. -

Chapter 19 the Chemistry of Aldehydes and Ketones. Addition Reactions
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 19 The Chemistry of Aldehydes and Ketones. Addition Reactions Solutions to In-Text Problems 19.1 (b) (d) (e) (g) 19.2 (a) 2-Propanone (d) (E)-3-Ethoxy-2-propenal (f) 4,4-Dimethyl-2,5-cyclohexadienone 19.3 (b) 2-Cyclohexenone has a lower carbonyl stretching frequency because its two double bonds are conjugated. 19.4 (b) The compound is 2-butanone: (c) The high frequency of the carbonyl absorption suggests a strained ring. (See Eq. 19.4, text p. 897.) In fact, cyclobutanone matches the IR stretching frequency perfectly and the NMR fits as well: 19.6 The structure and CMR assignments of 2-ethylbutanal are shown below. The two methyl groups are chemically equivalent, and the two methylene groups are chemically equivalent; all carbons with different CMR chemical shifts are chemically nonequivalent. INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 19 2 19.7 (a) The double bonds in 2-cyclohexenone are conjugated, but the double bonds in 3-cyclohexenone are not. Consequently, 2-cyclohexenone has the UV spectrum with the greater lmax. 19.9 Compound A, vanillin, should have a p T p* absorption at a greater lmax when dissolved in NaOH solution because the resulting phenolate can delocalize into the carboxaldehyde group; the resulting phenolate from compound B, isovanillin, on the other hand, can only delocalize in the aromatic ring. 19.11 The mass spectrum of 2-heptanone should have major peaks at m/z = 43 (from a-cleavage), 71 (from inductive cleavage), and 58 (from McLafferty rearrangement).