Soln-2212-PT3-S20-Ch-16-18.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Guide to the Nomenclature of Organic Chemistry
1 Brief Guide to the Nomenclature of Table 1: Components of the substitutive name Organic Chemistry (4S,5E)-4,6-dichlorohept-5-en-2-one for K.-H. Hellwich (Germany), R. M. Hartshorn (New Zealand), CH3 Cl O A. Yerin (Russia), T. Damhus (Denmark), A. T. Hutton (South 4 2 Africa). E-mail: [email protected] Sponsoring body: Cl 6 CH 5 3 IUPAC Division of Chemical Nomenclature and Structure suffix for principal hept(a) parent (heptane) one Representation. characteristic group en(e) unsaturation ending chloro substituent prefix 1 INTRODUCTION di multiplicative prefix S E stereodescriptors CHEMISTRY The universal adoption of an agreed nomenclature is a key tool for 2 4 5 6 locants ( ) enclosing marks efficient communication in the chemical sciences, in industry and Multiplicative prefixes (Table 2) are used when more than one for regulations associated with import/export or health and safety. fragment of a particular kind is present in a structure. Which kind of REPRESENTATION The International Union of Pure and Applied Chemistry (IUPAC) multiplicative prefix is used depends on the complexity of the provides recommendations on many aspects of nomenclature.1 The APPLIED corresponding fragment – e.g. trichloro, but tris(chloromethyl). basics of organic nomenclature are summarized here, and there are companion documents on the nomenclature of inorganic2 and Table 2: Multiplicative prefixes for simple/complicated entities polymer3 chemistry, with hyperlinks to original documents. An No. Simple Complicated No. Simple Complicated AND overall -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

Aldehydes and Ketones (Pp. 916-920)
Aldehydes and Ketones Klein, D. (2012). Aldehydes and Ketones. En Organic Chemistry (pp. 916-920). USA: Wiley. Espacio de Formación Multimodal 20.2 Nomenclature 917 916 CHAPTER 20 Aldehydes and Ketones 20.2 Nomenclature DO YOU REMEMBER? Before you go on, be sure you understand the following topics. If necessary, review the suggested sections Nomenclature of Aldehydes to prepare for this chapter: Recall that four discrete steps are required to name most classes of organic compounds (as we UÊ À}>À`ÊÀi>}iÌÃÊ-iVÌʣΰȮÊÊ UÊ ,iÌÀÃÞÌ iÌVÊ>>ÞÃÃÊ-iVÌÊ£Ó°x® saw with alkanes, alkenes, alkynes, and alcohols): UÊ "Ý`>ÌÊvÊ>V ÃÊ-iVÌʣΰ£ä® 1. Identify and name the parent. 6ÃÌÊÜÜÜ°ÜiÞ«ÕðVÊÌÊV iVÊÞÕÀÊÕ`iÀÃÌ>`}Ê>`ÊvÀÊÛ>Õ>LiÊ«À>VÌVi° Ê 2. Identify and name the substituents. 3. Assign a locant to each substituent. 4. Assemble the substituents alphabetically. Aldehydes are also named using the same four-step procedure. When applying this procedure 20.1 Introduction to Aldehydes and Ketones for naming aldehydes, the following guidelines should be followed: When naming the parent, the suffix “-al” indicates the presence of an aldehyde group: Aldehydes (RCHO) and ketones (R2CO) are similar in structure in that both classes of com- pounds possess a CO bond, called a carbonyl group: O Carbonyl Group H Butane Butanal O O When choosing the parent of an aldehyde, identify the longest chain that includes the carbon RH RR atom of the aldehydic group: An aldehyde A ketone The parent The carbonyl group of an aldehyde is flanked by a hydrogen atom, while the carbonyl group of must include HO a ketone is flanked by two carbon atoms. -
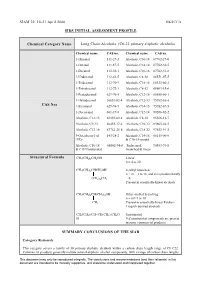
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Transcription 12.01.12
Lecture 2B • 01/12/12 We covered three different reactions for converting alcohols into leaving groups. One was to turn an alcohol into an alkyl chloride, that was using thionyl chloride. Second reaction was using tosyl chloride; the primary difference between those two reactions is one of stereochemistry. An inversion of stereochemistry does occur if you use thionyl chloride, because it does affect the carbon-oxygen bond, but because forming a tosylate does not touch the carbon-oxygen bond, only the oxygen- hydrogen bond, there’s no change in stereochemistry there. We then saw phosphorus tribromide that reacts very similarly to the thionyl chloride; you get an alkyl bromide instead, but it also has inversion of configuration. The last reaction is not a new mechanism, it is just an Sn2 reaction, it’s called the Finkelstein reaction. Really this works, in a sense, off of Le Châtelier’s principle. In solution, in theory, sodium iodide can displace bromide, but sodium bromide can displace iodide, so you can have an Sn2 reaction that goes back and forth and back and forth and back and forth. Except, sodium iodide is somewhat soluble in acetone, while sodium chloride and sodium bromide are not. So, in fact, one of the things that we get out of this as a by-product is sodium bromide, which, again, is not soluble in acetone and therefore precipitates out and is no longer part of the reaction mixture, so there’s no reverse reaction possible. Because of this solubility trick, it allows this reaction to be pulled forward, which means you can get the alkyl iodide. -

Reactions of Alcohols & Ethers 1
REACTIONS OF ALCOHOLS & ETHERS 1. Combustion (Extreme Oxidation) alcohol + oxygen carbon dioxide + water 2 CH3CH2OH + 6 O2 4 CO2 + 6 H2O 2. Elimination (Dehydration) ° alcohol H 2 S O 4/ 1 0 0 C alkene + water H2SO4/100 ° C CH3CH2CH2OH CH3CH=CH2 + H2O 3. Condensation ° excess alcohol H 2 S O 4 / 1 4 0 C ether + water H2SO4/140 ° C 2 CH3CH2OH CH3CH2OCH2CH3 + H2O 4. Substitution Lucas Reagent alcohol + hydrogen halide Z n C l2 alkyl halide + water ZnCl2 CH3CH2OH + HCl CH3CH2Cl + H2O • This reaction with the Lucas Reagent (ZnCl2) is a qualitative test for the different types of alcohols because the rate of the reaction differs greatly for a primary, secondary and tertiary alcohol. • The difference in rates is due to the solubility of the resulting alkyl halides • Tertiary Alcohol→ turns cloudy immediately (the alkyl halide is not soluble in water and precipitates out) • Secondary Alcohol → turns cloudy after 5 minutes • Primary Alcohol → takes much longer than 5 minutes to turn cloudy 5. Oxidation • Uses an oxidizing agent such as potassium permanganate (KMnO4) or potassium dichromate (K2Cr2O7). • This reaction can also be used as a qualitative test for the different types of alcohols because there is a distinct colour change. dichromate → chromium 3+ (orange) → (green) permanganate → manganese (IV) oxide (purple) → (brown) Tertiary Alcohol not oxidized under normal conditions CH3 KMnO4 H3C C OH NO REACTION K2Cr2O7 CH3 tertbutyl alcohol Secondary Alcohol ketone + hydrogen ions H O KMnO4 H3C C CH3 + K2Cr2O7 C + 2 H H3C CH3 OH propanone 2-propanol Primary Alcohol aldehyde + water carboxylic acid + hydrogen ions O KMnO4 O H H KMnO H H 4 + CH3CH2CH2OH C C C H C C C H + 2 H K2Cr2O7 + H2O K Cr O H H H 2 2 7 HO H H 1-propanol propanal propanoic acid 6. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Nomenclature of Steroids
Pure&App/. Chern.,Vol. 61, No. 10, pp. 1783-1822,1989. Printed in Great Britain. @ 1989 IUPAC INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY and INTERNATIONAL UNION OF BIOCHEMISTRY JOINT COMMISSION ON BIOCHEMICAL NOMENCLATURE* NOMENCLATURE OF STEROIDS (Recommendations 1989) Prepared for publication by G. P. MOSS Queen Mary College, Mile End Road, London El 4NS, UK *Membership of the Commission (JCBN) during 1987-89 is as follows: Chairman: J. F. G. Vliegenthart (Netherlands); Secretary: A. Cornish-Bowden (UK); Members: J. R. Bull (RSA); M. A. Chester (Sweden); C. LiCbecq (Belgium, representing the IUB Committee of Editors of Biochemical Journals); J. Reedijk (Netherlands); P. Venetianer (Hungary); Associate Members: G. P. Moss (UK); J. C. Rigg (Netherlands). Additional contributors to the formulation of these recommendations: Nomenclature Committee of ZUB(NC-ZUB) (those additional to JCBN): H. Bielka (GDR); C. R. Cantor (USA); H. B. F. Dixon (UK); P. Karlson (FRG); K. L. Loening (USA); W. Saenger (FRG); N. Sharon (Israel); E. J. van Lenten (USA); S. F. Velick (USA); E. C. Webb (Australia). Membership of Expert Panel: P. Karlson (FRG, Convener); J. R. Bull (RSA); K. Engel (FRG); J. Fried (USA); H. W. Kircher (USA); K. L. Loening (USA); G. P. Moss (UK); G. Popjiik (USA); M. R. Uskokovic (USA). Correspondence on these recommendations should be addressed to Dr. G. P. Moss at the above address or to any member of the Commission. Republication of this report is permitted without the need for formal IUPAC permission on condition that an acknowledgement, with full reference together with IUPAC copyright symbol (01989 IUPAC), is printed. -

Chapter 19 the Chemistry of Aldehydes and Ketones. Addition Reactions
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 19 The Chemistry of Aldehydes and Ketones. Addition Reactions Solutions to In-Text Problems 19.1 (b) (d) (e) (g) 19.2 (a) 2-Propanone (d) (E)-3-Ethoxy-2-propenal (f) 4,4-Dimethyl-2,5-cyclohexadienone 19.3 (b) 2-Cyclohexenone has a lower carbonyl stretching frequency because its two double bonds are conjugated. 19.4 (b) The compound is 2-butanone: (c) The high frequency of the carbonyl absorption suggests a strained ring. (See Eq. 19.4, text p. 897.) In fact, cyclobutanone matches the IR stretching frequency perfectly and the NMR fits as well: 19.6 The structure and CMR assignments of 2-ethylbutanal are shown below. The two methyl groups are chemically equivalent, and the two methylene groups are chemically equivalent; all carbons with different CMR chemical shifts are chemically nonequivalent. INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 19 2 19.7 (a) The double bonds in 2-cyclohexenone are conjugated, but the double bonds in 3-cyclohexenone are not. Consequently, 2-cyclohexenone has the UV spectrum with the greater lmax. 19.9 Compound A, vanillin, should have a p T p* absorption at a greater lmax when dissolved in NaOH solution because the resulting phenolate can delocalize into the carboxaldehyde group; the resulting phenolate from compound B, isovanillin, on the other hand, can only delocalize in the aromatic ring. 19.11 The mass spectrum of 2-heptanone should have major peaks at m/z = 43 (from a-cleavage), 71 (from inductive cleavage), and 58 (from McLafferty rearrangement). -

Naming Organic Chemicals Naming Organic Chemicals
B11CC3B17CA Naming Organic Chemicals B17CA Naming Organic Chemicals Naming Alkenes Alkenes are named in the same way that alkanes are named except that you use -ene as the stem ending. A comprehensive set of nomenclature rules has been devised by the International Union of Pure and The parent chain for the name is the longest chain containing the alkene group. Applied Chemistry (IUPAC). This sheet explains basic chemical nomenclature. Use a locant to indicate the position of the alkene and number from the end of the chain that gives the lowest value to this locant. Naming Alkanes 5 8 Straight chain alkanes are named with a stem to indicate the number of carbons in the chain followed CH2CH2CH2CH3 by the ending -ane. 1 ALKANE CH3CH2CH2CHCH3 4-methyloctane 4 methane CH4 hexane CH3CH2CH2CH2CH2CH3 ethane CH3CH3 heptane CH3CH2CH2CH2CH2CH2CH3 5 3 2 8 propane CH CH CH octane CH CH CH CH CH CH CH CH 3 2 3 3 2 2 2 2 2 2 3 CH2CH CHCH3 butane CH3CH2CH2CH3 nonane CH3CH2CH2CH2CH2CH2CH2CH2CH3 8 pentane CH CH CH CH CH decane CH CH CH CH CH CH CH CH CH CH ALKENE CH3CH2CH2CHCH3 5-methyloct-2-ene 3 2 2 2 3 3 2 2 2 2 2 2 2 2 3 5 For branched alkanes choose the longest chain as the parent chain for the name and include the side locant indicates position of double bond chains as substituent prefixes: methyl-, ethyl-, propyl-, butyl- etc. stem ending -ene 5 8 Notice that the locant indicating the position of the double bond is located BEFORE the ending 1 CH2CH2CH2CH3 4-methyloctane ‘-ene’. -

Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and Tert-Amyl Alcohol: a Density Functional Study
Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and tert-Amyl Alcohol: A Density Functional Study Wolfgang Forner¨ and Hassan M. Badawi Department of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia Reprint requests to Wolfgang Forner.¨ E-mail: [email protected] Z. Naturforsch. 2013, 68b, 841 – 851 / DOI: 10.5560/ZNB.2013-3003 Received February 9, 2013 We have calculated the vibrational spectra of isoamyl alcohol and of tert-amyl alcohol using Den- sity Functional Theory (DFT) with the Becke-3 Lee Yang Parr (B3LYP) functional and a 6-311+G** atomic basis set. The energies of the conformers were also calculated with ab initio Perturbation Theory of second (MP2) and fourth order restricted to single, double and quadruple excitations (MP4 SDQ) with the same basis set. We found rather complicated equilibria of four conformations in each case, counting only those with appreciable abundancies. PED data were compared with GAUSSVIEW animations. The calculated wavenumbers agree rather well with the experimental ones when the gauche-trans conformer is assumed as the most important one for isoamyl alcohol, and the gauche- gauche one for tert-amyl alcohol. However, some of the experimental bands had to be assigned also to other conformers, indicating their presence in the equilibrium mixture. Due to sterical reasons both the CO and the OH bonds appear to be weaker in the tertiary alcohol, considering the wavenumbers of the CO and OH bond stretching vibrations. The bond lengths point into the same direction, however, the OH bond in the tertiary alcohol is only slightly longer than that in the primary alcohol. -

Advantages of Nodal Numbering for Uniquely Identifying Atoms in Chemical Nomenclature Alan L
CROATICA CHEMICA ACTA CCACAA 59 (3) 547-563 (198G)1 YU ISSN 0011-1643 CCA-1668 UDC 541 Original Scientific Paper Advantages of Nodal Numbering for Uniquely Identifying Atoms in Chemical Nomenclature Alan L. Goodson 1060 Woodmere Road, CoLumbus, Ohio 43220, U.S.A. and Noel Lozac'h Institut des Sciences de la Matiere et du Rayonnement, Universite de Caen, 14032 Caen Cedex, France Received September 2, 1985 There are situations in chemical nomenclature where duplica- tion of locants creates difficulties (a) in describing modifications to chemical structures, (b) in describing stereochemistry, (c) in identifying isotopically-labeled atoms, and (d) in uniquely iden- tifying atoms for crystallography, Nodal nomenclature is shown to avoid these problems and to simplify such descriptions. INTRODUCTION The first concerted international agreements on chemical nornenclature- were made in Geneva in 18921. From these agreements has aris en the Inter- national Union of Pure and Applied Chemistry (IUPAC), as we know it today. IUPAC has, in general, »confined its efforts to codifying sound practices. which already existed-". IUPAC has published a set of internationally-agreed rules of organic chemistry", However, more than one sound practice is some- times described (e. g., for naming organic compounds containing phosphorus,. arsenic, antimony or bismuth) without any indication of which practice is preferred. For practical reasons, indexers of chemical literature, such as Chemical Abstracts Service and Beilstein, must be more selective, i. e. more systematic, than IUP AC and must even develop their own rules when IUP AC rules are vague or non-existent. The rules published by IUP AC include the powerful and commonly-used technique of substitutive nomenclature.