Glossary of Terms Used in Physical Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Grade Level 10 Units 1-15
COPPELL ISD SUBJECT YEAR AT A GLANCE GRADE HEMISTRY UNITS C LEVEL 1-15 10 Program Transfer Goals ● Ask questions, recognize and define problems, and propose solutions. ● Safely and ethically collect, analyze, and evaluate appropriate data. ● Utilize, create, and analyze models to understand the world. ● Make valid claims and informed decisions based on scientific evidence. ● Effectively communicate scientific reasoning to a target audience. PACING 1st 9 Weeks 2nd 9 Weeks 3rd 9 Weeks 4th 9 Weeks Unit 1 Unit 2 Unit 3 Unit 4 Unit 5 Unit 6 Unit Unit Unit Unit Unit Unit Unit Unit Unit 7 8 9 10 11 12 13 14 15 1.5 wks 2 wks 1.5 wks 2 wks 3 wks 5.5 wks 1.5 2 2.5 2 wks 2 2 2 wks 1.5 1.5 wks wks wks wks wks wks wks Assurances for a Guaranteed and Viable Curriculum Adherence to this scope and sequence affords every member of the learning community clarity on the knowledge and skills on which each learner should demonstrate proficiency. In order to deliver a guaranteed and viable curriculum, our team commits to and ensures the following understandings: Shared Accountability: Responding -

Prebiological Evolution and the Metabolic Origins of Life
Prebiological Evolution and the Andrew J. Pratt* Metabolic Origins of Life University of Canterbury Keywords Abiogenesis, origin of life, metabolism, hydrothermal, iron Abstract The chemoton model of cells posits three subsystems: metabolism, compartmentalization, and information. A specific model for the prebiological evolution of a reproducing system with rudimentary versions of these three interdependent subsystems is presented. This is based on the initial emergence and reproduction of autocatalytic networks in hydrothermal microcompartments containing iron sulfide. The driving force for life was catalysis of the dissipation of the intrinsic redox gradient of the planet. The codependence of life on iron and phosphate provides chemical constraints on the ordering of prebiological evolution. The initial protometabolism was based on positive feedback loops associated with in situ carbon fixation in which the initial protometabolites modified the catalytic capacity and mobility of metal-based catalysts, especially iron-sulfur centers. A number of selection mechanisms, including catalytic efficiency and specificity, hydrolytic stability, and selective solubilization, are proposed as key determinants for autocatalytic reproduction exploited in protometabolic evolution. This evolutionary process led from autocatalytic networks within preexisting compartments to discrete, reproducing, mobile vesicular protocells with the capacity to use soluble sugar phosphates and hence the opportunity to develop nucleic acids. Fidelity of information transfer in the reproduction of these increasingly complex autocatalytic networks is a key selection pressure in prebiological evolution that eventually leads to the selection of nucleic acids as a digital information subsystem and hence the emergence of fully functional chemotons capable of Darwinian evolution. 1 Introduction: Chemoton Subsystems and Evolutionary Pathways Living cells are autocatalytic entities that harness redox energy via the selective catalysis of biochemical transformations. -
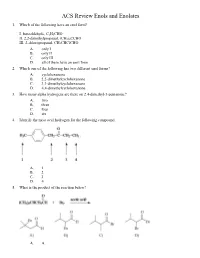
Ochem ACS Review 18 Enols and Enolates
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

A Facile Approach to Prepare Multiple Heteroatom-Doped Carbon
www.nature.com/scientificreports OPEN A Facile Approach to Prepare Multiple Heteroatom-Doped Carbon Materials from Imine- Received: 17 November 2017 Accepted: 19 February 2018 Linked Porous Organic Polymers Published: xx xx xxxx Juan Yang, Min Xu, Jingyu Wang , Shangbin Jin & Bien Tan In this paper, we proposed a new strategy to prepare multiple heteroatom doped (N, P-doped) porous carbon materials with high surface area of ~1,535 m2 g−1 simply by pyrolysis of imine-linked porous organic polymers (POPs) synthesized via Schif base condensation. The strategy is simple without any post-processing and various heteroatoms could be involved. Scanning electron microscopy, Raman spectra, Nitrogen gas adsorption-desorption, X-ray photoelectron spectroscopy have been used to characterize the morphology, the structure and the composition of the materials. The multiple heteroatom doped porous carbon materials also display high electrocatalytic performance as exampled by the application in oxygen reduction, which showed the catalyst favors 4-electron transfer during the process, along with superior stability and higher tolerance to methanol as compared to the Pt/C. These results indicate the present method is promising for the preparation of multi-heteroatom doped carbon materials in the application of electrocatalysis. Porous organic polymers (POPs) are a type of organic polymers with large surface area, tunable skeleton and high stability1,2. Tey have attracted extensive attentions for broad applications in the felds of gas storage and separations, catalysis and energy storage3–5. Tere are many methodologies reported for the preparation of porous polymers6–10. Among them, Schif base reaction is one of the most versatile ways due to the facile, one-pot, catalyst-free, quantitative synthesis11. -

An Analysis of Electrophilic Aromatic Substitution: a “Complex Approach” PCCP
Volume 23 Number 9 7 March 2021 Pages 5033–5682 PCCP Physical Chemistry Chemical Physics rsc.li/pccp ISSN 1463-9076 PERSPECTIVE Janez Cerkovnik et al . An analysis of electrophilic aromatic substitution: a “complex approach” PCCP View Article Online PERSPECTIVE View Journal | View Issue An analysis of electrophilic aromatic substitution: a ‘‘complex approach’’† Cite this: Phys. Chem. Chem. Phys., a a b 2021, 23, 5051 Nikola Stamenkovic´, Natasˇa Poklar Ulrih and Janez Cerkovnik * Electrophilic aromatic substitution (EAS) is one of the most widely researched transforms in synthetic organic chemistry. Numerous studies have been carried out to provide an understanding of the nature of its reactivity pattern. There is now a need for a concise and general, but detailed and up-to-date, overview. The basic principles behind EAS are essential to our understanding of what the mechanisms underlying EAS are. To date, textbook overviews of EAS have provided little information about the Received 5th October 2020, mechanistic pathways and chemical species involved. In this review, the aim is to gather and present the Accepted 21st December 2020 up-to-date information relating to reactivity in EAS, with the implication that some of the key concepts DOI: 10.1039/d0cp05245k will be discussed in a scientifically concise manner. In addition, the information presented herein suggests certain new possibilities to advance EAS theory, with particular emphasis on the role of modern Creative Commons Attribution-NonCommercial 3.0 Unported Licence. rsc.li/pccp -

Opportunities for Catalysis in the 21St Century
Opportunities for Catalysis in The 21st Century A Report from the Basic Energy Sciences Advisory Committee BASIC ENERGY SCIENCES ADVISORY COMMITTEE SUBPANEL WORKSHOP REPORT Opportunities for Catalysis in the 21st Century May 14-16, 2002 Workshop Chair Professor J. M. White University of Texas Writing Group Chair Professor John Bercaw California Institute of Technology This page is intentionally left blank. Contents Executive Summary........................................................................................... v A Grand Challenge....................................................................................................... v The Present Opportunity .............................................................................................. v The Importance of Catalysis Science to DOE.............................................................. vi A Recommendation for Increased Federal Investment in Catalysis Research............. vi I. Introduction................................................................................................ 1 A. Background, Structure, and Organization of the Workshop .................................. 1 B. Recent Advances in Experimental and Theoretical Methods ................................ 1 C. The Grand Challenge ............................................................................................. 2 D. Enabling Approaches for Progress in Catalysis ..................................................... 3 E. Consensus Observations and Recommendations.................................................. -

FIRE-SAFE POLYMERS and POLYMER COMPOSITES September 2004 6
DOT/FAA/AR-04/11 Fire-Safe Polymers and Polymer Office of Aviation Research Washington, D.C. 20591 Composites September 2004 Final Report This document is available to the U.S. public through the National Technical Information Service (NTIS), Springfield, Virginia 22161. U.S. Department of Transportation Federal Aviation Administration NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents or use thereof. The United States Government does not endorse products or manufacturers. Trade or manufacturer's names appear herein solely because they are considered essential to the objective of this report. This document does not constitute FAA certification policy. Consult your local FAA aircraft certification office as to its use. This report is available at the Federal Aviation Administration William J. Hughes Technical Center’s Full-Text Technical Reports page: actlibrary.act.faa.gov in Adobe Acrobat portable document format (PDF). Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AR-04/11 4. Title and Subtitle 5. Report Date FIRE-SAFE POLYMERS AND POLYMER COMPOSITES September 2004 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Huiqing Zhang 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) Polymer Science and Engineering University of Massachusetts Amhurst, MA 01003 11. Contract or Grant No. 12. Sponsoring Agency Name and Address 13. Type of Report and Period Covered U.S. Department of Transportation Federal Aviation Administration Office of Aviation Research 14. -

Dehydrogenation by Heterogeneous Catalysts
Dehydrogenation by Heterogeneous Catalysts Daniel E. Resasco School of Chemical Engineering and Materials Science University of Oklahoma Encyclopedia of Catalysis January, 2000 1. INTRODUCTION Catalytic dehydrogenation of alkanes is an endothermic reaction, which occurs with an increase in the number of moles and can be represented by the expression Alkane ! Olefin + Hydrogen This reaction cannot be carried out thermally because it is highly unfavorable compared to the cracking of the hydrocarbon, since the C-C bond strength (about 246 kJ/mol) is much lower than that of the C-H bond (about 363 kJ/mol). However, in the presence of a suitable catalyst, dehydrogenation can be carried out with minimal C-C bond rupture. The strong C-H bond is a closed-shell σ orbital that can be activated by oxide or metal catalysts. Oxides can activate the C-H bond via hydrogen abstraction because they can form O-H bonds, which can have strengths comparable to that of the C- H bond. By contrast, metals cannot accomplish the hydrogen abstraction because the M- H bonds are much weaker than the C-H bond. However, the sum of the M-H and M-C bond strengths can exceed the C-H bond strength, making the process thermodynamically possible. In this case, the reaction is thought to proceed via a three centered transition state, which can be described as a metal atom inserting into the C-H bond. The C-H bond bridges across the metal atom until it breaks, followed by the formation of the corresponding M-H and M-C bonds.1 Therefore, dehydrogenation of alkanes can be carried out on oxides as well as on metal catalysts. -
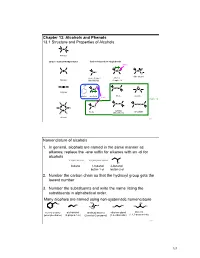
Chapter 13.Pptx
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Electronic Structures and Spin Topologies of #-Picoliniumyl Radicals
Subscriber access provided by UNIV OF MISSOURI COLUMBIA Article Electronic Structures and Spin Topologies of #-Picoliniumyl Radicals. A Study of the Homolysis of N-Methyl-#-picolinium and of Benzo-, Dibenzo-, and Naphthoannulated Analogs Rainer Glaser, Yongqiang Sui, Ujjal Sarkar, and Kent S. Gates J. Phys. Chem. A, 2008, 112 (21), 4800-4814 • DOI: 10.1021/jp8011987 • Publication Date (Web): 22 May 2008 Downloaded from http://pubs.acs.org on December 12, 2008 More About This Article Additional resources and features associated with this article are available within the HTML version: • Supporting Information • Access to high resolution figures • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article The Journal of Physical Chemistry A is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 4800 J. Phys. Chem. A 2008, 112, 4800–4814 Electronic Structures and Spin Topologies of γ-Picoliniumyl Radicals. A Study of the Homolysis of N-Methyl-γ-picolinium and of Benzo-, Dibenzo-, and Naphthoannulated Analogs Rainer Glaser,*,† Yongqiang Sui,† Ujjal Sarkar,† and Kent S. Gates*,†,‡ Departments of Chemistry and Biochemistry, UniVersity of Missouri-Columbia, Columbia, Missouri 65211 ReceiVed: February 9, 2008 Radicals resulting from one-electron reduction of (N-methylpyridinium-4-yl) methyl esters have been reported to yield (N-methylpyridinium-4-yl) methyl radical, or N-methyl-γ-picoliniumyl for short, by heterolytic cleavage of carboxylate. This new reaction could provide the foundation for a new structural class of bioreductively activated, hypoxia-selective antitumor agents. N-methyl-γ-picoliniumyl radicals are likely to damage DNA by way of H-abstraction and it is of paramount significance to assess their H-abstraction capabilities.