An Analysis of Electrophilic Aromatic Substitution: a “Complex Approach” PCCP
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Planar Cyclopenten‐4‐Yl Cations: Highly Delocalized Π Aromatics
Angewandte Research Articles Chemie How to cite: Angew.Chem. Int. Ed. 2020, 59,18809–18815 Carbocations International Edition: doi.org/10.1002/anie.202009644 German Edition: doi.org/10.1002/ange.202009644 Planar Cyclopenten-4-yl Cations:Highly Delocalized p Aromatics Stabilized by Hyperconjugation Samuel Nees,Thomas Kupfer,Alexander Hofmann, and Holger Braunschweig* 1 B Abstract: Theoretical studies predicted the planar cyclopenten- being energetically favored by 18.8 kcalmolÀ over 1 (MP3/ 4-yl cation to be aclassical carbocation, and the highest-energy 6-31G**).[11–13] Thebishomoaromatic structure 1B itself is + 1 isomer of C5H7 .Hence,its existence has not been verified about 6–14 kcalmolÀ lower in energy (depending on the level experimentally so far.Wewere now able to isolate two stable of theory) than the classical planar structure 1C,making the derivatives of the cyclopenten-4-yl cation by reaction of bulky cyclopenten-4-yl cation (1C)the least favorable isomer.Early R alanes Cp AlBr2 with AlBr3.Elucidation of their (electronic) solvolysis studies are consistent with these findings,with structures by X-raydiffraction and quantum chemistry studies allylic 1A being the only observable isomer, notwithstanding revealed planar geometries and strong hyperconjugation the nature of the studied cyclopenteneprecursor.[14–18] Thus, interactions primarily from the C Al s bonds to the empty p attempts to generate isomer 1C,orits homoaromatic analog À orbital of the cationic sp2 carbon center.Aclose inspection of 1B,bysolvolysis of 4-Br/OTs-cyclopentene -

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -

Nucleophilic Aromatic Substitution on Aryl-Amido Ligands Promoted by Oxidizing Osmium(IV) Centers
Inorg. Chem. 2004, 43, 5804−5815 Nucleophilic Aromatic Substitution on Aryl-Amido Ligands Promoted by Oxidizing Osmium(IV) Centers Jake D. Soper, Erik Saganic, David Weinberg, David A. Hrovat, Jason B. Benedict,† Werner Kaminsky,† and James M. Mayer* Department of Chemistry, Campus Box 351700, UniVersity of Washington, Seattle, Washington 98195-1700 Received January 30, 2004 IV Addition of amine nucleophiles to acetonitrile solutions of the Os anilido complex TpOs(NHPh)Cl2 (1) [Tp ) hydrotris(1-pyrazolyl)borate] gives products with derivatized anilido ligands, i.e., TpOs[NH-p-C6H4(N(CH2)5)]Cl2 (2) from piperidine and TpOs[NH-p-C6H4N(CH2)4]Cl2 (3) from pyrrolidine. These materials are formed in ∼30% yield III under anaerobic conditions, together with ∼60% yields of the Os aniline complex TpOs(NH2Ph)Cl2 (5). Formation of the para-substituted materials 2 or 3 from 1 involves oxidative removal of two hydrogen atoms (two H+ and two - e ). The oxidation can be accomplished by 1, forming 5,orbyO2. Related reactions have been observed with other amines and with the 2-naphthylamido derivative, which gives an ortho-substituted product. Kinetic studies indicate an addition−elimination mechanism involving initial attack of the amine nucleophile on the anilido ligand. These are unusual examples of nucleophilic aromatic substitution of hydrogen. Ab initio calculations on 1 show that the LUMO has significant density at the ortho and para positions of the anilido ligand, resembling the LUMO of nitrobenzene. By analogy with nucleophilic aromatic substitution, 2 is quantitatively formed from piperidine and IV the p-chloroanilide TpOs(NH-p-C6H4Cl)Cl2 (7). -

Intermolecular Forces
Intermolecular Forces The intermolecular forces between molecules are important in the properties of all solid and liquid materials. They are key to reactions that take place in biological molecules. Proteins form their secondary and tertiary structures through hydrogen-bonding and London forces. DNA forms because of hydrogen bonding between base pairs. Enzymes function when molecules interact with the protein active site in these biological catalysts. Outline • Types of Intermolecular Forces • Entropy Considerations • Intermolecular Forces and DNA • Homework Types of Intermolecular Forces Solutions consist of a solvent and solute. There are gas, liquid, and solid solutions but in this unit we are concerned with liquids. The solvent then is a liquid phase molecular material that makes up most of the solution. Water is a good example of a solvent. The solute is a smaller quantity of anything that is dissolved in the solvent. It can be a gas (O2 in water), a liquid (water dissolved in ethanol), or a solid (NaCl dissolved in water). In any solution, the molecules or ions of the solute are randomly distributed among the molecules of the solvent. What are the intermolecular forces? They are listed in the table below along with covalent and ionic bonding for comparison. Notice that they have different dependence on the distance between the attracting particles, r. Materials dissolve in a solution when there are strong intermolecular forces between the solute and the solvent. London Dispersion Forces We could discount intermolecular interactions between gas-phase molecules because these molecules are mostly far apart and moving rapidly relative to each other. In the liquid phases, all molecules interact with one another. -

Changes: Physical Or Chemical? by Cindy Grigg
Changes: Physical or Chemical? By Cindy Grigg 1 If you have studied atoms, you know that atoms are the building blocks of matter. Atoms are so small they cannot be seen with an ordinary microscope. Yet atoms make up everything in the universe. Atoms can combine with different atoms and make new substances. Substances can also break apart into separate atoms. These changes are called chemical changes or reactions. Chemical reactions happen when atoms gain, lose, or share electrons. What about when water freezes into ice? Do you think that's a chemical change? 2 When water freezes, it has changed states. You probably already know about the four states of matter. They are solid, liquid, gas, and plasma. Plasma is the fourth state of matter and is the most common state in the universe. However, it is rarely found on Earth. Plasma occurs as ball lightning and in stars. Water is a common substance that everyone has seen in its three states of matter. Water in its solid state is called ice. Water in the liquid state is just called water. Water as a gas is called water vapor. We can easily cause water to change states by changing its temperature. Water will freeze at 32 degrees Fahrenheit (0 Celsius). However, no chemical change has occurred. The atoms have not combined or broken apart to make a different substance; it is still water or H2O. When we heat water to a temperature of 212 F. or 100 Celsius, it will change into a gas called water vapor. Changes in states of matter are just physical changes. -

How Does Argumentation-Based Instruction Affect Pre-Service Science Teachers’ Conceptual Understanding of Organic Chemistry? the Case of Aromatic Compounds1
Online Science Education Journal http://dergipark.gov.tr/ofed 2020; 5(1): 32 - 51. How Does Argumentation-based Instruction Affect Pre-service Science Teachers’ Conceptual Understanding of Organic Chemistry? The Case of Aromatic Compounds1 Gülten Şendur*, Dokuz Eylul University, Buca Faculty of Education, Department of Mathematics and Science Education, Chemistry Education Division, Izmir, Turkey Ezgi Kurt, Dokuz Eylul University, Institute of Educational Sciences, Izmir, Turkey Büşra Hekimoğlu, Ministry of National Education, Istanbul, Turkey *Corresponding Author: [email protected] Şendur, G., Kurt, E. & Hekimoğlu, B. (2020). How does argumentation-based instruction affect pre-service science teachers’ conceptual understanding of organic To cite this article chemistry? The case of aromatic compounds. Online Science Education Journal, 5(1): 32-51. Article Info Abstract Article History Aromatic compounds are one of the fundamental topics in Organic Chemistry. For this reason, creating learning environments that will contribute to pre-service Received: teachers’ meaningful understanding of aromatic compounds is of importance. The 04 May 2020 purpose of this study was to explore whether argumentation-based instruction has an effect on the conceptual understanding of pre-service science teachers in the Accepted: topic of aromatic compounds. In pursuit of this aim, the study was conducted in 05 June 2020 quasi-experimental, pre-test/post-test and control group design during the 2016- 2017 academic year at the Science Education Division of a state university in Keywords Turkey. Two classes were randomly selected as an experimental group (N=30) and a control group (N=35). The data collection instruments used in the study Argumentation Aromatic were pre- and post-tests, consisting of 10 open-ended questions. -

Metal Carbonyls
MODULE 1: METAL CARBONYLS Key words: Carbon monoxide; transition metal complexes; ligand substitution reactions; mononuclear carbonyls; dinuclear carbonyls; polynuclear carbonyls; catalytic activity; Monsanto process; Collman’s reagent; effective atomic number; 18-electron rule V. D. Bhatt / Selected topics in coordination chemistry / 2 MODULE 1: METAL CARBONYLS LECTURE #1 1. INTRODUCTION: Justus von Liebig attempted initial experiments on reaction of carbon monoxide with metals in 1834. However, it was demonstrated later that the compound he claimed to be potassium carbonyl was not a metal carbonyl at all. After the synthesis of [PtCl2(CO)2] and [PtCl2(CO)]2 reported by Schutzenberger (1868) followed by [Ni(CO)4] reported by Mond et al (1890), Hieber prepared numerous compounds containing metal and carbon monoxide. Compounds having at least one bond between carbon and metal are known as organometallic compounds. Metal carbonyls are the transition metal complexes of carbon monoxide containing metal-carbon bond. Lone pair of electrons are available on both carbon and oxygen atoms of carbon monoxide ligand. However, as the carbon atoms donate electrons to the metal, these complexes are named as carbonyls. A variety of such complexes such as mono nuclear, poly nuclear, homoleptic and mixed ligand are known. These compounds are widely studied due to industrial importance, catalytic properties and structural interest. V. D. Bhatt / Selected topics in coordination chemistry / 3 Carbon monoxide is one of the most important π- acceptor ligand. Because of its π- acidity, carbon monoxide can stabilize zero formal oxidation state of metals in carbonyl complexes. 2. SYNTHESIS OF METAL CARBONYLS Following are some of the general methods of preparation of metal carbonyls. -

Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2
Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2 RED Metal-ligand and metal-metal bonding of the transition metal elements Synopsis Lecture 1: Trends of the transition metal series. Ionic vs Covalent bonding. Nomenclature. Electron counting. Lecture 2: Thermodynamics of complex formation. Why complexes form. Recap of molecular orbital theory. 18-electron rule. Lecture 3: Ligand classes. -donor complexes. Octahedral ML6 molecular orbital energy diagram. Lecture 3: - acceptor ligands and synergic bonding. Binding of CO, CN , N2, O2 and NO. Lecture 4: Alkenes, M(H2) vs M(H)2, Mn(O2) complexes, PR3. Lecture 5: 2- - 2- 3- donor ligands, metal-ligand multiple bonds, O , R2N , RN , N . Lecture 6: ML6 molecular orbital energy diagrams incorporating acceptor and donor ligands. Electron counting revisited and link to spectrochemical series. Lecture 7: Kinetics of complex formation. Substitution mechanisms of inorganic complexes. Isomerisation. Lecture 8: Ligand effects on substitution rates (trans-effect, trans-influence). Metal and geometry effects on substitution rates. Lecture 9: Outer sphere electron transfer. Lecture 10: Inner Sphere electron transfer. Bridging ligands. 2 Learning Objectives: by the end of the course you should be able to i) Use common nomenclature in transition metal chemistry. ii) Count valence electrons and determine metal oxidation state in transition metal complexes. iii) Understand the physical basis of the 18-electron rule. iv) Appreciate the synergic nature of bonding in metal carbonyl complexes. v) Understand the relationship between CO, the 'classic' -acceptor and related ligands such as NO, CN, N2, and alkenes. 2 vi) Describe the nature of the interaction between -bound diatomic molecules (H2, O2) and their relationship to -acceptor ligands. -
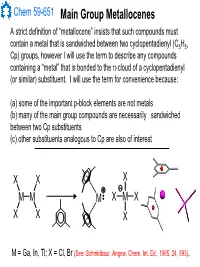
Main Group Metallocenes
Chem 59-651 Main Group Metallocenes A strict definition of “metallocene” insists that such compounds must contain a metal that is sandwiched between two cyclopentadienyl (C5H5, Cp) groups, however I will use the term to describe any compounds containing a “metal” that is bonded to the π-cloud of a cyclopentadienyl (or similar) substituent. I will use the term for convenience because: (a) some of the important p-block elements are not metals (b) many of the main group compounds are necessarily sandwiched between two Cp substituents (c) other substituents analogous to Cp are also of interest XX X MM M XMX X X X M = Ga, In, Tl; X = Cl, Br (See: Schmidbaur, Angew. Chem. Int. Ed., 1985, 24, 893). Chem 59-651 Cyclopentadienyl Ligand Basics The structural features and bonding interactions between cyclopentadienyl groups and metal atoms shows considerable variation and is described using a few different conventions (See Jutzi and Burford, Chem. Rev., 1999, 99, 969 and references therein). In most main group metallocenes, even those that are σ-bonded in the solid state, the rings rotate rapidly through a series of 1,2-shifts. A Cp group can be considered to be p-bonded to an element if the Hapticity: element sits inside the cylinder defined by the 5 carbon atoms of M MM the Cp ligand. The center of mass of the 5 C atoms is known as the centroid. η1-Cp η2-Cp η3-Cp M M M A σ-bonded Cp can be identified η4-Cp η5-Cp ≡η5-Cp by the angle at the ipso carbon M and bond lengths should indicate single bonds to the ipso carbon and a localized diene structure for σ the α and β carbon fragment. -

Application of the Covalent Bond Classification Method for The
Article pubs.acs.org/jchemeduc Application of the Covalent Bond Classification Method for the Teaching of Inorganic Chemistry Malcolm L. H. Green† and Gerard Parkin*,‡ † Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, United Kingdom ‡ Department of Chemistry, Columbia University, New York, New York 10027, United States *S Supporting Information ABSTRACT: The Covalent Bond Classification (CBC) method provides a means to classify covalent molecules according to the number and types of bonds that surround an atom of interest. This approach is based on an elementary molecular orbital analysis of the bonding involving the central atom (M), with the various interactions being classified according to the number of electrons that each neutral ligand contributes to the bonding orbital. Thus, with respect to the atom of interest (M), the ligand can contribute either two (L), one (X), or zero (Z) electrons to a bonding orbital. A normal covalent bond is represented as M−X, whereas dative covalent bonds are represented as either M←LorM→Z, according to whether the ligand is the donor (L) fi or acceptor (Z). A molecule is classi ed as [MLlXxZz] according to the number of L, X, and Z ligand functions that surround M. Not only does fi fi the [MLlXxZz] designation provide a formal classi cation of a molecule, but it also indicates the electron con guration, the valence, and the number of nonbonding electrons on M. As such, the classification allows a student to understand relationships between molecules, thereby increasing their ability to conceptualize and learn the chemistry of the elements.