Radical Approaches to Alangium and Mitragyna Alkaloids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

"Alcohol Activation" That Would Be Stereochemically Complementary to That Involving Reaction of an Alcohol with P / S Halides (Notes of Nov 20)
CHEM 203 Topics Discussed on Nov. 23 Desirability of a method for "alcohol activation" that would be stereochemically complementary to that involving reaction of an alcohol with P / S halides (notes of Nov 20): PBr3 CH S Na 3 overall (inversion of H (inversion) H retention configuration) Br SMe (S)-2-bromobutane (R)-configured pdt. H OH (R)-2-butanol ? (S)-configured pdt. overall H inversion SMe Sulfonyl chlorides: para-toluenesulfonyl ("tosyl") chloride, methanesulfonyl ("mesyl") chloride O O O R S Cl H3C S Cl S Cl O O O A generic sulfonyl methanesulfonyl chloride Toluene para-toluenesulfonyl chloride chloride: R = any ( "mesyl chloride" ) (= methyl benzene) ( "tosyl chloride" ) alkyl group Pyridine: a weakly basic, nucleophilic analog of benzene in which an N atom replaces a CH unit: pyridine N Reaction of primary and secondary alcohols with sulfonyl chlorides in the presence of pyridine: formation of sulfonate esters (= alkyl sulfonates): R1 O R1 O OH + Cl S R N O S R + R2 O R2 N O H Cl a generic primary or an "alkyl sulfonate" secondary alcohol ("tosylate", "mesylate," etc.) note: tertiary alcohols are insufficiently nucleophilic to react with sulfonyl chlorides Presumed mechanism for the formation of sulfonate esters from primary and secondary (but not tertiary) alcohols and sulfonyl chlorides: • slow rate of reaction of an alcohol with sulfonyl chlorides in the presence of generic bases Lecture of Nov. 23 p. 2 • pyridine as a nucleophilic catalyst that greatly accelerates the reaction of an alcohol with a sulfonyl chloride by: (i) -

Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide
Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry at the Massachusetts Institute of Technology February, 1999 @1999 Jinhua Song All rights Reserved The author hereby grants MIT permissions to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part. Signature of Author: Department of Chemistry September 25, 1998 Certified by: Professor Satoru Masamune A. C. Cope Professor of Chemistry Thesis Supervisor Accepted by:, ProfessotDietmar Seyferth, Chairman Departmental Committee on Graduate Students MASSACHUSETTS INSTITUTE OF TECHNOLOGY LrL J This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Timothy M. Swager Chairman Professor Satoru Masamune Thesis Supervisor Professor Rick L. Danheiser , 2 Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry on September 25, 1998, in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry Abstract Presented are the stereoselective syntheses of the A (C18-C28), B (C14-C17), C (C6-C13), D (Cl-C5), C'D' (C1-C13) fragments and the efficient coupling of B and C'D' fragments of the marine natural product miyakolide, a 24-membered polyketide macrolide which exhibits anti-cancer activity. Fragment A was synthesized from the chiral aldehyde 4-4 through the successful application of the newly developed boron mediated anti-selective aldol methodology using the chiral ester 3-4. -

The Synthesis and Applications of N-Alkenyl Aziridines
The Synthesis and Applications of N-Alkenyl Aziridines by Nicholas A. Afagh A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Nicholas A. Afagh 2010 The Synthesis and Applications of N-Alkenyl Aziridines Nicholas A. Afagh Master of Science Department of Chemistry University of Toronto 2010 Abstract N-alkenyl aziridines are a unique class of molecules that do not behave as typical enamines as a result of the inability of the nitrogen atom lone-pair of electrons to delocalize. The attenuated nucleophilicity of these enamines presents opportunities for the selective functionalization and reactivity not available to classical enamines. An operationally simple and mild copper-mediated coupling has been developed that facilitates the preparation of a broad range of N-alkenyl aziridines not available through existing methods. The preparation and reactivity of highly- functionalized N-alkenyl aziridines are reported. Also reported is the application of the chemoselective amine/aldehyde/alkyne (A 3) multicomponent coupling involving amphoteric aziridine aldehydes as the aldehyde component. This coupling allows access to propargyl amines with pendent aziridine functionality. ii Acknowledgments First and foremost, I would like to thank my supervisor, Professor Andrei K. Yudin for his continuous support and encouragement over the past two years. His wealth of knowledge and profound insight into all matters chemistry made for many interesting discussions. In addition, I would like to thank all the members of the Yudin group past and present with whom I have had the distinct pleasure of working alongside and shared many late evenings. -

ECO-Ssls for Pahs
Ecological Soil Screening Levels for Polycyclic Aromatic Hydrocarbons (PAHs) Interim Final OSWER Directive 9285.7-78 U.S. Environmental Protection Agency Office of Solid Waste and Emergency Response 1200 Pennsylvania Avenue, N.W. Washington, DC 20460 June 2007 This page intentionally left blank TABLE OF CONTENTS 1.0 INTRODUCTION .......................................................1 2.0 SUMMARY OF ECO-SSLs FOR PAHs......................................1 3.0 ECO-SSL FOR TERRESTRIAL PLANTS....................................4 5.0 ECO-SSL FOR AVIAN WILDLIFE.........................................8 6.0 ECO-SSL FOR MAMMALIAN WILDLIFE..................................8 6.1 Mammalian TRV ...................................................8 6.2 Estimation of Dose and Calculation of the Eco-SSL ........................9 7.0 REFERENCES .........................................................16 7.1 General PAH References ............................................16 7.2 References Used for Derivation of Plant and Soil Invertebrate Eco-SSLs ......17 7.3 References Rejected for Use in Derivation of Plant and Soil Invertebrate Eco-SSLs ...............................................................18 7.4 References Used in Derivation of Wildlife TRVs .........................25 7.5 References Rejected for Use in Derivation of Wildlife TRV ................28 i LIST OF TABLES Table 2.1 PAH Eco-SSLs (mg/kg dry weight in soil) ..............................4 Table 3.1 Plant Toxicity Data - PAHs ..........................................5 Table 4.1 -
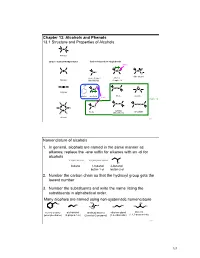
Chapter 13.Pptx
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Alfa Olefins Cas N
OECD SIDS ALFA OLEFINS FOREWORD INTRODUCTION ALFA OLEFINS CAS N°:592-41-6, 111-66-0, 872-05-9, 112-41-4, 1120-36-1 UNEP PUBLICATIONS 1 OECD SIDS ALFA OLEFINS SIDS Initial Assessment Report For 11th SIAM (Orlando, Florida, United States 1/01) Chemical Name: 1-hexene Chemical Name: 1-octene CAS No.: 592-41-6 CAS No.: 111-66-0 Chemical Name: 1-decene Chemical Name: 1-dodecene CAS No.: 872-05-9 CAS No.: 112-41-4 Chemical Name: 1-tetradecene CAS No.: 1120-36-1 Sponsor Country: United States National SIDS Contract Point in Sponsor Country: United States: Dr. Oscar Hernandez Environmental Protection Agency OPPT/RAD (7403) 401 M Street, S.W. Washington, DC 20460 Sponsor Country: Finland (for 1-decene) National SIDS Contact Point in Sponsor Country: Ms. Jaana Heiskanen Finnish Environment Agency Chemicals Division P.O. Box 140 00251 Helsinki HISTORY: SIDS Dossier and Testing Plan were reviewed at the SIDS Review Meeting or in SIDS Review Process on October 1993. The following SIDS Testing Plan was agreed: No testing ( ) Testing (x) Combined reproductive/developmental on 1-hexene, combined repeat dose/reproductive/developmental on 1-tetradecene and acute fish, daphnid and algae on 1- tetradecene. COMMENTS: The following comments were made at SIAM 6 and have been incorporated in this version of the SIAR: 2 UNEP PUBLICATIONS OECD SIDS ALFA OLEFINS 1. The use of QSAR calculations for aquatic toxicity, 2. More quantitative assessment of effects; and 3. Provide more details for each endpoint. The following comments were made at SIAM 6, but were not incorporated into the SIAR for the reasons provided: 1. -

Synthesis and Kinetics of Novel Ionic Liquid Soluble Hydrogen Atom Transfer Reagents
Synthesis and kinetics of novel ionic liquid soluble hydrogen atom transfer reagents Thomas William Garrard Submitted in total fulfilment of the requirements of the degree Doctor of Philosophy June 2018 School of Chemistry The University of Melbourne Produced on archival quality paper ORCID: 0000-0002-2987-0937 Abstract The use of radical methodologies has been greatly developed in the last 50 years, and in an effort to continue this progress, the reactivity of radical reactions in greener alternative solvents is desired. The work herein describes the synthesis of novel hydrogen atom transfer reagents for use in radical chemistry, along with a comparison of rate constants and Arrhenius parameters. Two tertiary thiol-based hydrogen atom transfer reagents, 3-(6-mercapto-6-methylheptyl)-1,2- dimethyl-3H-imidazolium tetrafluoroborate and 2-methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol, have been synthesised. These are modelled on traditional thiol reagents, with a six-carbon chain with an imidazole ring on one end and tertiary thiol on the other. 3-(6-mercapto-6-methylheptyl)- 1,2-dimethyl-3H-imidazolium tetrafluoroborate comprises of a charged imidazolium ring, while 2- methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol has an uncharged imidazole ring in order to probe the impact of salt formation on radical kinetics. The key step in the synthesis was addition of thioacetic acid across an alkene to generate a tertiary thioester, before deprotection with either LiAlH4 or aqueous NH3. Arrhenius plots were generated to give information on rate constants for H-atom transfer to a primary alkyl radical, the 5-hexenyl radical, in ethylmethylimidazolium bis(trifluoromethane)sulfonimide. -

O/C -O-O-( X, Generally Carried out at a Temperature in the Range of 20 and (B) Mineral Acid Salts of These Compounds
3,256,288 United States Patent Office Patented June 14, 1966 W 2 -Cl, -Br or -SONH2, with an appropriate imino ether 3,256,288 hydrochloride having the formula -SUBSTITUTED AMNOALKYL-2-ARYLOXY METHYLEBENZRADAZOLE COMPOUNDS E Clarence L. Moyle, Care, and Diomed M. Cher, Mid land, Mich., assignors to The Dow Chemical Company, Midland, Mich., a corporation of Delaware W (III) No Drawing. Fied May 24, 1962, Ser. No. 197,285 wherein in this and succeeding formulas, E is -H, -R, 9 Claims. (C. 260-294.7) -CI, -Br, -OH, -OR or -CONH2 and R' is a lower alkyl group, to produce the desired benzimidazole product This invention is directed to benzimidazole compounds, O and R'OH, NH3 and HCl by-products. The gaseous NH particularly (a) N-substituted benzimidazole compounds and HCl generally evolve from the reaction mixture al having the formula though some of the HC1 may react with NH and remain in the reaction mixture as ammonium chloride salt or may react with the basic benzimidazole product and remain 5 as the hydrochloride salt thereof. / N Y In carrying out the preparation, substantially equimolar proportions of the reactants are employed although either reactant may be employed in excess. The reaction is O/C -o-o-( x, generally carried out at a temperature in the range of 20 and (b) mineral acid salts of these compounds. In this from 60 to 82° C. for a period of from about 20 to 72 and succeeding formulas-NR'R'' is di(lower-alkyl)ami hours. It is preferred that an alcoholic solvent be em no, piperidino, morpholino or pyrrolidino; X is -H, ployed in this process. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Hydrophilic Thin Coating and Method of Manufacturing the Same
Europaisches Patentamt European Patent Office © Publication number: 0 599 150 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION © Application number: 93118306.5 int. CIA B05D 1/18, C03C 17/30, C08J 7/04 @ Date of filing: 11.11.93 ® Priority: 12.11.92 JP 302124/92 © Applicant: MATSUSHITA ELECTRIC INDUSTRIAL Co., Ltd. @ Date of publication of application: 1006-banchi, Oaza-Kadoma 01.06.94 Bulletin 94/22 Kadoma-shi, Osaka(JP) © Designated Contracting States: @ Inventor: Ohtake, Tadashi DE FR GB Kawakitanaka-machi 30-15 Neyagawa-shi, Osaka 572(JP) Inventor: Mino, Norihisa Senrioka Higashi 4-6-8-806 Settsu-shi, Osaka 566(JP) Inventor: Ogawa, Kazufumi Aoyama 2-3-50 Nara-shi, Nara 630(JP) © Representative: VOSSIUS & PARTNER Siebertstrasse 4 D-81675 Munchen (DE) © Hydrophilic thin coating and method of manufacturing the same. © The invention relates to a hydrophilic thin film and a method of manufacturing the same in which a hydrophilic thin film 4 is formed by incorporating or fixing molecules comprising hydrophilic groups to a chemically adsorbed film on the surface of a substrate 1 . 7\ oj— Cl O I OH 2 ^4 $ -0— Si — 0 Si — 0 Si- -o- -Si-0- I I I I 0 0 0 0 — > 7> , , , 1 , , r-i-. i ///////////// X FIG . Rank Xerox (UK) Business Services (3. 10/3.09/3.3.4) EP 0 599 150 A1 The invention relates to a hydrophilic thin film and a method of manufacturing the same. More specifically, the invention relates to a hydrophilic thin film and its method of manufacture, in which molecules comprising hydrophilic groups are incorporated or chemically bonded to the surface of a chemically adsorbed film on a substrate surface. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive.