Radical Cyclization in Heterocycle Synthesis. 12.1) Sulfanyl Radical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Synthesis and Kinetics of Novel Ionic Liquid Soluble Hydrogen Atom Transfer Reagents
Synthesis and kinetics of novel ionic liquid soluble hydrogen atom transfer reagents Thomas William Garrard Submitted in total fulfilment of the requirements of the degree Doctor of Philosophy June 2018 School of Chemistry The University of Melbourne Produced on archival quality paper ORCID: 0000-0002-2987-0937 Abstract The use of radical methodologies has been greatly developed in the last 50 years, and in an effort to continue this progress, the reactivity of radical reactions in greener alternative solvents is desired. The work herein describes the synthesis of novel hydrogen atom transfer reagents for use in radical chemistry, along with a comparison of rate constants and Arrhenius parameters. Two tertiary thiol-based hydrogen atom transfer reagents, 3-(6-mercapto-6-methylheptyl)-1,2- dimethyl-3H-imidazolium tetrafluoroborate and 2-methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol, have been synthesised. These are modelled on traditional thiol reagents, with a six-carbon chain with an imidazole ring on one end and tertiary thiol on the other. 3-(6-mercapto-6-methylheptyl)- 1,2-dimethyl-3H-imidazolium tetrafluoroborate comprises of a charged imidazolium ring, while 2- methyl-7-(2-methylimidazol-1-yl)heptane-2-thiol has an uncharged imidazole ring in order to probe the impact of salt formation on radical kinetics. The key step in the synthesis was addition of thioacetic acid across an alkene to generate a tertiary thioester, before deprotection with either LiAlH4 or aqueous NH3. Arrhenius plots were generated to give information on rate constants for H-atom transfer to a primary alkyl radical, the 5-hexenyl radical, in ethylmethylimidazolium bis(trifluoromethane)sulfonimide. -

O/C -O-O-( X, Generally Carried out at a Temperature in the Range of 20 and (B) Mineral Acid Salts of These Compounds
3,256,288 United States Patent Office Patented June 14, 1966 W 2 -Cl, -Br or -SONH2, with an appropriate imino ether 3,256,288 hydrochloride having the formula -SUBSTITUTED AMNOALKYL-2-ARYLOXY METHYLEBENZRADAZOLE COMPOUNDS E Clarence L. Moyle, Care, and Diomed M. Cher, Mid land, Mich., assignors to The Dow Chemical Company, Midland, Mich., a corporation of Delaware W (III) No Drawing. Fied May 24, 1962, Ser. No. 197,285 wherein in this and succeeding formulas, E is -H, -R, 9 Claims. (C. 260-294.7) -CI, -Br, -OH, -OR or -CONH2 and R' is a lower alkyl group, to produce the desired benzimidazole product This invention is directed to benzimidazole compounds, O and R'OH, NH3 and HCl by-products. The gaseous NH particularly (a) N-substituted benzimidazole compounds and HCl generally evolve from the reaction mixture al having the formula though some of the HC1 may react with NH and remain in the reaction mixture as ammonium chloride salt or may react with the basic benzimidazole product and remain 5 as the hydrochloride salt thereof. / N Y In carrying out the preparation, substantially equimolar proportions of the reactants are employed although either reactant may be employed in excess. The reaction is O/C -o-o-( x, generally carried out at a temperature in the range of 20 and (b) mineral acid salts of these compounds. In this from 60 to 82° C. for a period of from about 20 to 72 and succeeding formulas-NR'R'' is di(lower-alkyl)ami hours. It is preferred that an alcoholic solvent be em no, piperidino, morpholino or pyrrolidino; X is -H, ployed in this process. -
![[(4-Chlorophenyl) Sulfanyl] Ethoxy- 3-Methoxy-5-[5-(3,4,5-Trimethoxyphenyl)-2-Furyl]Benzonitrile](https://docslib.b-cdn.net/cover/2932/4-chlorophenyl-sulfanyl-ethoxy-3-methoxy-5-5-3-4-5-trimethoxyphenyl-2-furyl-benzonitrile-892932.webp)
[(4-Chlorophenyl) Sulfanyl] Ethoxy- 3-Methoxy-5-[5-(3,4,5-Trimethoxyphenyl)-2-Furyl]Benzonitrile
Available online a t www.derpharmachemica.com Scholars Research Library Der Pharma Chemica, 2015, 7(3):242-247 (http://derpharmachemica.com/archive.html) ISSN 0975-413X CODEN (USA): PCHHAX Synthesis and antibacterial activity of 2-2-[(4-chlorophenyl) sulfanyl] ethoxy- 3-methoxy-5-[5-(3,4,5-trimethoxyphenyl)-2-furyl]benzonitrile P. Veerabhadra Swamy*a,b , K. B. Chandrasekhar c and Pullaiah China Kambhampati a aLaxai Avanti Life Sciences, Lab#9, ICICI Knowledge park, Shameerpet, Turkapally Village, Hyderabad bDepartment of Chemistry, Jawaharlal Nehru Technological University, Hyderabad, Hyderabad, Telengana, India cDepartment of Chemistry, Jawaharlal Nehru Technological University, Anantapur, Anantapuramu, A.P., India _____________________________________________________________________________________________ ABSTRACT The present paper describes the synthesis and of (2-(4-chlorophenylthio)ethoxy)-3-methoxy-5-(5-(3,4,5- trimethoxyphenyl)furan-2-yl)benzonitrile from commercially available vanillin and 3,4,5-trimethoxy acetophenone as starting materials utilizing green reagents and solvents. The antibacterial test results indicated that the title compound displayed excellent activity against both Gram-positive bacteria (Staphylococcus aureus and Bacillus cereus) and Gram-negative bacteria: (Escherichia Coli and Pseudomonas aeruginosa). Keywords: Antibacterial activity, Furan, 3,4,5-trimethoxy acetophenone, vanillin, synthesis _____________________________________________________________________________________________ INTRODUCTION Furan derivatives -

United States Patent (19) 11) Patent Number: 4,731,479 Bod Et Al
United States Patent (19) 11) Patent Number: 4,731,479 Bod et al. 45 Date of Patent: Mar. 15, 1988 (54) N-SULFAMYL-3-HALOPROPIONAMIDINES Attorney, Agent, or Firm-Karl F. Ross; Herbert Dubno; 75 Inventors: Péter Bod, Gyömrö; Kálmán Jonathan Myers Harsányi, Budapest; Eva Againée (57) ABSTRACT Csongor, Budapest; Erik Bogsch, Budapest; Eva Fekecs, Budapest; The invention relates to new propionamidine deriva Ferenc Trischler, Budapest; György tives of formula (I) Domány, Budapest; István Szabadkai, Budapest; Béla Hegedis, Budapest, NH2HX (I) all of Hungary x^-sn-so- NH2 73) Assignee: Richter Gedeon Vegyeszeti Gyar Rt., Budapest, Hungary wherein (21) Appl. No.: 905,833 X is halogen, and to a process for their preparation. According to the 22 Filed: Sep. 10, 1986 invention compounds of the formula (I) are prepared by 30 Foreign Application Priority Data reacting a 3-halopropionitrile of the formula (III) Sep. 11, 1985 HU) Hungary .............................. 3423/85 51) Int. Cl." ............................................ CO7C 143/72 (III) 52 U.S.C. ...................................................... 564/79 58 Field of Search .......................................... 564/79 wherein (56) References Cited X is as defined above, U.S. PATENT DOCUMENTS with sulfamide of the formula (II) 3,121,084 2/1964 Winberg................................ 564/79 (II) FOREIGN PATENT DOCUMENTS 905408 12/1986 Belgium. NH-i-NH. O OTHER PUBLICATIONS Wagner et al, "Synthetic Organic Chemistry', (1953), in the presence of a hydrogen halide. p. 635. Compounds of the formula (I) are useful intermediates Richter, "Text Book of Organic Chemistry,” (1952), p. 216. in the preparation of famotidine. Primary Examiner-Anton H. Sutto 1 Claim, No Drawings 4,731,479 1. -

Direct Electrophilic N-Trifluoromethylthiolation of Amines with Trifluoromethanesulfenamide
Supporting Information for Direct electrophilic N-trifluoromethylthiolation of amines with trifluoromethanesulfenamide Sébastien Alazet1,2, Kevin Ollivier1 and Thierry Billard*1,2 Address: 1Institute of Chemistry and Biochemistry (ICBMS – UMR CNRS 5246), Université de Lyon, Université Lyon 1, CNRS, 43 Bd du 11 novembre 1918 – 69622 Lyon, France and 2CERMEP - in vivo imaging, Groupement Hospitalier Est, 59 Bd Pinel – 69003 Lyon, France Email: Thierry Billard - [email protected] * Corresponding author Experimental procedure S1 Table of Contents General Information .............................................................................................................. S3 Typical procedure .................................................................................................................. S3 1-Phenyl-4-[(trifluoromethyl)sulfanyl]piperazine (3a) ...................................................................................... S3 1-Benzyl-4-[(trifluoromethyl)sulfanyl]piperazine (3b) ...................................................................................... S4 1-(2-Methoxyphenyl)-4-[(trifluoromethyl)sulfanyl]piperazine (3c) .................................................................. S4 1-(Pyridin-2-yl)-4-[(trifluoromethyl)sulfanyl]piperazine (3d) ........................................................................... S4 tert-Butyl 4-[(trifluoromethyl)sulfanyl]piperazine-1-carboxylate (3e) .............................................................. S5 2,2,6,6-Tetramethyl-1-[(trifluoromethyl)sulfanyl]piperidine -

Polymer Additives, Contaminants and Non-Intentionally Added Substances in Consumer Products: Combined Migration, Permeation and Toxicity Analyses in Skin
Polymer additives, contaminants and non-intentionally added substances in consumer products: Combined migration, permeation and toxicity analyses in skin Inaugural-Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin by Nastasia Bartsch from Gräfelfing, Munich 2018 This thesis was carried out at the Federal Institute for Risk Assessment (BfR) in Berlin from May 2013 to February 2017 under the supervision of Prof. Dr. Dr. Andreas Luch. 1st Reviewer: Prof. Dr. Dr. Andreas Luch 2nd Reviewer: Prof. Dr. Daniel Klinger Date of defense: 14 December 2018 ERKLÄRUNG Hiermit versichere ich, die vorliegende Dissertation mit dem Titel „Polymer additives, contaminants and non-intentionally added substances in consumer products: Combined migration, permeation and toxicity analyses in skin“ selbstständig und ohne Benutzung anderer als der zugelassenen Hilfsmittel angefertigt zu haben. Alle angeführten Zitate sind als solche kenntlich gemacht. Die vorliegende Arbeit wurde in keinem früheren Promotionsverfahren angenommen oder als ungenügend beurteilt. Berlin, den 25. September 2018 Nastasia Bartsch Die Dissertation wurde in englischer Sprache verfasst. III Danksagung Ich möchte meinen Dank auf diesem Wege an Herrn Prof. Dr. Dr. Luch richten, für die Möglichkeit, meine Doktorarbeit in der Abteilung Chemikalien- und Produktsicherheit am Bundesinstitut für Risikobewertung anzufertigen. Ich schätze sehr, dass Professor Luch stets meine interdisziplinäre Herangehensweise an das Thema gefördert hat, mir Raum gab, meine Ideen umzusetzen und mich darin bestärkte, meine Arbeit in einem breiten wissenschaftlichen Kontext zu betrachten. Herrn Prof. Dr. Klinger gilt mein Dank für sein Interesse an meinem Projekt und für die Übernahme der Zweitgutachterschaft. -
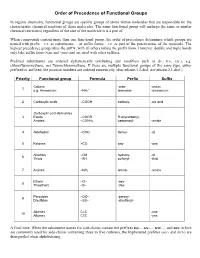
Priority of Functional Groups
Order of Precedence of Functional Groups In organic chemistry, functional groups are specific groups of atoms within molecules that are responsible for the characteristic chemical reactions of those molecules. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. When compounds contain more than one functional group, the order of precedence determines which groups are named with prefix – i.e. as substituents –, or suffix forms – i.e. as part of the parent name of the molecule. The highest precedence group takes the suffix, with all others taking the prefix form. However, double and triple bonds only take suffix form (-ene and -yne) and are used with other suffixes. Prefixed substituents are ordered alphabetically (excluding any modifiers such as di-, tri-, etc.), e.g. chlorofluoromethane, not fluorochloromethane. If there are multiple functional groups of the same type, either prefixed or suffixed, the position numbers are ordered numerically (thus ethane-1,2-diol, not ethane-2,1-diol.) Priority Functional group Formula Prefix Suffix Cations -onio- -onium 1 + e.g. Ammonium –NH4 ammonio- -ammonium 2 Carboxylic acids –COOH carboxy- -oic acid Carboxylic acid derivatives 3 Esters –COOR R-oxycarbonyl- Amides –CONH2 carbamoyl- -amide 4 Aldehydes –CHO formyl- -al 5 Ketones >CO oxo- -one Alcohols –OH hydroxy- -ol 6 Thiols –SH sulfanyl- -thiol 7 Amines –NH2 amino- -amine Ethers –O– -oxy- 8 Thioethers –S– -thio- Peroxides –OO– -peroxy- 9 Disulfides –SS– -disulfanyl- Alkenes C=C -ene 10 Alkynes C≡C -yne A final note: When the substituent names for side-chains contain the prefixes iso..., sec-..., tert-..., and neo (which are commonly used for side-chains containing three to five carbons), the hyphenated prefixes (sec- and tert-) are disregarded when alphabetizing. -

Migration Reactions
CHAPTER 3 Radical Reactions: Part 1 A. J. CLARK, J. V. GEDEN, and N. P. MURPHY Department of Chemistry, University of Warwick Introduction Rearrangements Group Migration β-Scission (Ring Opening) Ring Expansion Intramolecular Addition Cyclization Tandem Reactions Radical Annulation Fragmentation, Recombination, and Homolysis Atom Abstraction Reactions Hydrogen abstraction by Carbon-centred Radicals Hydrogen Abstraction by Heteroatom-centred Radicals Halogen Abstraction Halogenation Addition Reactions Addition to Carbon-Carbon Multiple Bonds Addition to Oxygen-containing Multiple Bonds Addition to Nitrogen-containing Multiple Bonds 1 Homolytic Substitution Aromatic Substitution SH2 and Related Reactions Reactivity Effects Polarity and Philicity Stability of Radicals Stereoselectivity in Radical Reactions Stereoselectivity in Cyclization Stereoselectivity in Addition Reactions Stereoselectivity in Atom Transfer Redox Reactions Radical Ions Anion Radicals Cation Radicals Peroxides, Peroxyl, and Hydroxyl Radicals Peroxides Peroxyl Radicals Hydroxyl Radical References 2 Introduction Free radical chemistry continues to be a focus for research with a number of reviews being published in 2000. Green aspects of chemistry have attracted a lot of attention with most work conducted investigating atmospheric chemistry, however on a review the use of supercritical fluids in radical reactions dealing solvent effects on chemical reactivity has appeared.1A The kinetics and thermochemistry of a range of free radical reactions has been reviewed. The review includes descriptions of the kinetics of a variety of unimolecular decomposition and isomerisation reactions as well as bimolecular hydrogen atom abstraction, additions, combinations and disproportionations. 204. In synthetic applications a review on the hydroxylation of benzene to phenol using Fenton’s reagent has appeared 254 as well as reviews on the synthesis of heterocycles using SRN1 mechansims279, and radical reactions controlled by Lewis Acids, 265. -

The Kinetics of Oxygen and SO2 Consumption by Red Wines. What Do They Tell About Oxidation Mechanisms and About Changes in Wine Composition?
Accepted Manuscript The kinetics of oxygen and SO2 consumption by red wines. What do they tell about oxidation mechanisms and about changes in wine composition? Vanesa Carrascón, Anna Vallverdú-Queralt, Emmanuelle Meudec, Nicolas Sommerer, Purificación Fernandez-Zurbano, Vicente Ferreira PII: S0308-8146(17)31425-5 DOI: http://dx.doi.org/10.1016/j.foodchem.2017.08.090 Reference: FOCH 21639 To appear in: Food Chemistry Received Date: 19 March 2017 Revised Date: 27 August 2017 Accepted Date: 28 August 2017 Please cite this article as: Carrascón, V., Vallverdú-Queralt, A., Meudec, E., Sommerer, N., Fernandez-Zurbano, P., Ferreira, V., The kinetics of oxygen and SO2 consumption by red wines. What do they tell about oxidation mechanisms and about changes in wine composition?, Food Chemistry (2017), doi: http://dx.doi.org/10.1016/ j.foodchem.2017.08.090 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 The kinetics of oxygen and SO2 consumption by red wines. What do they tell about oxidation mechanisms and about changes in wine composition? Vanesa Carrascón1, Anna Vallverdú-Queralt2, Emmanuelle Meudec2, Nicolas Sommerer2, Purificación Fernandez-Zurbano3, Vicente Ferreira1,3* 1 Laboratory for Aroma Analysis and Enology. Instituto Agroalimentario de Aragón (IA2- Unizar-CITA). -

LIST of PRODUCTS BULK DRUGS Sl. No Name of the Product Production TPM 1. Carvedilol Phosphate 2.50 2. Citalopram Hydrobromide 10
LIST OF PRODUCTS BULK DRUGS Sl. No Name of the product Production TPM 1. Carvedilol phosphate 2.50 2. Citalopram Hydrobromide 10.00 3. Closantel sodium 5.00 4. Esomeprazole magnesium trihydrate 1.50 5. Fexofinadine hydrochloride 2.00 6. Fosfomycin trometamol 3.00 7. Gaba-pentin 3.00 8. Itraconozole 2.00 9. Lansoprazole 3.00 10. Lornoxicam 4.00 11. Montelukast sodium 3.00 12. Omeprazole 5.00 13. Ondansetron 3.00 14. Oxyclozanide 5.00 15. Ritonavir 3.00 16. Rosuvastatin calcium 3.00 17. Setraline hydrochloride 2.00 18. Sparfloxacin 2.00 19. Terbinafine hydrochloride 3.00 Total 65.00 INTERMEDIATES Sl. No Name of the product Production TPM 1 Tri Chloro Salisylic Acid 3.00 2 5-AminO-2-Hydroxy Benzoic acid 2.00 3 5-Amino-2-dibenzylamino-1,6-diphenyl-hex-4-en-3-one 2.00 4 2-Amino-6-chloro-3-nitro pyridine 3.00 5 4-Chloro butyryl chloride 3.00 CHEMICALS Sl.No Name of the product Production TPM 1 Tributyl Tin chloride 90 2 L-Menthol 90 3 Dicyclo Hexylcarbomidiimide 90 SOLVENT RECOVERY Sl.No Name of the product Production TPM 1 All solvents 450 Industry proposes to produce • 2.0 TPD either from bulk drug group or intermediate group • 3.0 TPD from chemicals group • 15.0 TPD solvent recovery CARVEDILOL PHOSPHATE Carvedilol Phosphate can be manufactured in four stages. Stage-1: 1-(2-bromo ethoxy)-2-methoxy benzene and benzyl amine reacts together in the presence of toluene solvent medium forms stage-1 compound. O Toluene Br + NH2 O 1-(2-Bromo-ethoxy)- 2-methoxy-benzene Benzylamine C9H11BrO2 C7H9N Mol. -

Download Article (PDF)
126 Notizen I(E)-l,2-Difluoro-2-(pentafluoro- Discussion A6-sulfanyl)ethenyl|phosphonates We have found that organic phosphites (la and Holger Wessolowskia, Gerd-Volker lb) will react with l,2,2-trifluoro-l-(pentafluoro- >„6-sulfanyl)ethylene (2) [5] to give the first F5S Röschenthaler*-3, Rolf Winterb, and group containing alkenylphosphonates, 4 a, b, Gary L. G ard*b [1] which are moisture-sensitive colorless liquids a Institut für Anorganische und Physikalische (Scheme 1). The (E) isomers exclusively were found, Chemie der Universität Bremen, Leobener Straße, D-2800 Bremen 33, F.R.G. derived from the intermediates 3 a, b in which there b Department of Chemistry, Portland State is maximum repulsion between the phosphonium University, Portland, Oregon 97207, U.S.A. and sulfanyl groupings [4], The 70 eV El mass spec Z. Naturforsch. 46b, 126-128(1991); tra show no parent molecular ion, but for 4 a the ion received June 15, 1990 [M + 1]+ was found; this has been observed in a few cases, for similar compounds [ 6]. For 4b the 1,2,2-T rifluoro-1 -(pentafluoro-A6-sulfanyl)- ethylene, Silylated Phosphites, Phosphonates [M -CH 3]+ fragment was obtained in keeping with results reported for O-trimethylsilylated phos For the first time SF5 containing organic phos 3)2 phonates, SF5CF = CFP(0)(0R')2 have been phonates, e.g. for CF3CF = CFP(0)(0SiMe [7], prepared from trimethylsilylphosphites, In the IR spectra, substitution of one fluorine (R'0)2P0SiMe, (R1 = Et, R1 = SiMe,) and atom in 2 by the phosphonate groupings decreases trifluorovinylsulfur pentafluoride, SF5CF = CF2 considerably (as expected) the C=C stretching fre in an Arbuzov type reaction.