Conjugate Addition Or Direct Addition to the Carbonyl Group?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
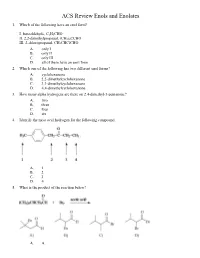
Ochem ACS Review 18 Enols and Enolates
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

SAFETY DATA SHEET Isopropyl Alcohol
SAFETY DATA SHEET Isopropyl Alcohol Section 1. Identification GHS product identifier : Isopropyl Alcohol Chemical name : Isopropyl alcohol Other means of : isopropanol; 2-Propanol identification Product type : Liquid. Product use : Synthetic/Analytical chemistry. Synonym : isopropanol; 2-Propanol SDS # : 001105 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 2 substance or mixture EYE IRRITATION - Category 2A SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : May form explosive mixtures with air. Highly flammable liquid and vapor. Causes serious eye irritation. May cause drowsiness or dizziness. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Prevention : Wear protective gloves. Wear eye or face protection. Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. Use explosion- proof electrical, ventilating, lighting and all material-handling equipment. Use only non- sparking tools. Take precautionary measures against static discharge. Keep container tightly closed. Use only outdoors or in a well-ventilated area. Avoid breathing vapor. Wash hands thoroughly after handling. Response : IF INHALED: Remove person to fresh air and keep comfortable for breathing. Call a POISON CENTER or physician if you feel unwell. -

United States Patent Office Patented Nov
3,221,026 United States Patent Office Patented Nov. 30, 1965 2 3,221,026 prepared by reaction of a dicyanoketene acetal of the SALTS OF 1,1-DCYANO-2,2,2-TRIALKOXY formula ETHANES Owen W. Webster, Wilmington, Del, assignor to E. I. du Pont de Nemours and Company, Wilmington, Del., a corporation of Delaware No Drawing. Filed Feb. 13, 1962, Ser. No. 172,875 wherein R2 and R3 have the meanings defined above in the 2 Claims. (C. 260-340.9) general formula for the products of this invention, with This invention relates to salts of polycyano compounds, one molar equivalent of an alkali metal alkoxide of an and more particularly, to salts of polycyanopolyalkoxy alcohol having 1-8 carbon atoms at a temperature below ethanes and a process for their preparation. 10° C., and preferably at a temperature between 0 and The salts are derivatives of tetracyanoethylene which -80° C., in the presence of an inert reaction medium, is a very reactive compound that has received considerable e.g., an excess of the alcohol from which the alkoxide is study during the last few years. A large number of new 5 derived, or an ether such as diethyl ether, dioxane, tetra and valuable compounds have been prepared from it, and hydrofuran, ethylene glycol dimethyl ether and the like. now a new class of polycyano compounds is provided by As in the case of the reaction starting with tetracyano the present invention. ethylene, the reaction mixture in this case should also The novel compounds of this invention are salts of the be anhydrous to obtain the best results. -

Synthesis of Novel Single-Source Precursors for CVD of Mixed-Metal Tungsten Oxide
Synthesis of novel single-source precursors for CVD of mixed-metal tungsten oxide Hamid Choujaa A thesis submitted for the degree of Doctor of Philosophy University of Bath Department of Chemistry March 2008 COPYRIGHT Attention is drawn to the fact that copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognize that its copyright rests with its author and that no quotation the thesis and no information derived from it may be published without the prior written consent of the author. This thesis may be made available for consultation within the University Library and may be photocopied or lent to other libraries for the purposes of consultation. TABLE OF CONTENTS Abstract ....................................................................................................................................... i Acknowledgements .................................................................................................................... iii Abbreviations and Acronyms .................................................................................................... iv 1. INTRODUCTION .................................................................................................................. 1 1.1 Generality about tungsten(VI) oxide ............................................................................. 1 1.1.1 The different lattice structures of tungsten oxide ........................................... 1 1.1.2 Electronic and -

8.6 Acidity of Alcohols and Thiols 355
08_BRCLoudon_pgs5-1.qxd 12/8/08 11:05 AM Page 355 8.6 ACIDITY OF ALCOHOLS AND THIOLS 355 ural barrier to the passage of ions. However, the hydrocarbon surface of nonactin allows it to enter readily into, and pass through, membranes. Because nonactin binds and thus transports ions, the ion balance crucial to proper cell function is upset, and the cell dies. Ion Channels Ion channels, or “ion gates,” provide passageways for ions into and out of cells. (Recall that ions are not soluble in membrane phospholipids.) The flow of ions is essen- tial for the transmission of nerve impulses and for other biological processes. A typical chan- nel is a large protein molecule imbedded in a cell membrane. Through various mechanisms, ion channels can be opened or closed to regulate the concentration of ions in the interior of the cell. Ions do not diffuse passively through an open channel; rather, an open channel contains regions that bind a specific ion. Such an ion is bound specifically within the channel at one side of the membrane and is somehow expelled from the channel on the other side. Remark- ably, the structures of the ion-binding regions of these channels have much in common with the structures of ionophores such as nonactin. The first X-ray crystal structure of a potassium- ion channel was determined in 1998 by a team of scientists at Rockefeller University led by Prof. Roderick MacKinnon (b. 1956), who shared the 2003 Nobel Prize in Chemistry for this work. The interior of the channel contains binding sites for two potassium ions; these sites are oxygen-rich, much like the interior of nonactin. -

N-BUTYL ALCOHOL
Right to Know Hazardous Substance Fact Sheet Common Name: n-BUTYL ALCOHOL Synonyms: Propyl Carbinol; n-Butanol CAS Number: 71-36-3 Chemical Name: 1-Butanol RTK Substance Number: 1330 Date: November 1998 Revision: January 2008 DOT Number: UN 1120 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE n-Butyl Alcohol is a colorless liquid with a strong, sweet Hazard Summary alcohol odor. It is used as a solvent for fats, waxes, shellacs, Hazard Rating NJDOH NFPA resins, gums, and varnish, in making hydraulic fluids, and in HEALTH - 2 medications for animals. FLAMMABILITY - 3 REACTIVITY - 0 f ODOR THRESHOLD = 1 to 15 ppm FLAMMABLE f Odor thresholds vary greatly. Do not rely on odor alone to POISONOUS GASES ARE PRODUCED IN FIRE determine potentially hazardous exposures. CONTAINERS MAY EXPLODE IN FIRE Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; Reasons for Citation 4=severe f n-Butyl Alcohol is on the Right to Know Hazardous f n-Butyl Alcohol can affect you when inhaled and by Substance List because it is cited by OSHA, ACGIH, DOT, passing through the skin. NIOSH, DEP, IRIS, NFPA and EPA. f Contact can irritate and burn the skin. f This chemical is on the Special Health Hazard Substance f n-Butyl Alcohol can irritate and burn the eyes with possible List. eye damage. f Inhaling n-Butyl Alcohol can irritate the nose, throat and lungs. f Exposure to n-Butyl Alcohol can cause headache, dizziness, nausea and vomiting. SEE GLOSSARY ON PAGE 5. f n-Butyl Alcohol can damage the liver, kidneys, hearing, and sense of balance. -

Nitric Oxide Activation Facilitated by Cooperative Multimetallic Electron Transfer Within an Iron- Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Nitric oxide activation facilitated by cooperative multimetallic electron transfer within an iron- Cite this: Chem. Sci.,2018,9,6379 – † All publication charges for this article functionalized polyoxovanadate alkoxide cluster have been paid for by the Royal Society a a a a b of Chemistry F. Li, R. L. Meyer, S. H. Carpenter, L. E. VanGelder, A. W. Nichols, C. W. Machan, b M. L. Neidiga and E. M. Matson *a A series of NO-bound, iron-functionalized polyoxovanadate–alkoxide (FePOV–alkoxide) clusters have been synthesized, providing insight into the role of multimetallic constructs in the coordination and activation of a substrate. Upon exposure of the heterometallic cluster to NO, the vanadium-oxide metalloligand is oxidized by a single electron, shuttling the reducing equivalent to the {FeNO} subunit to 7 V IV form a {FeNO} species. Four NO-bound clusters with electronic distributions ranging from [V3V2 ] Received 1st March 2018 {FeNO}7 to [VIV]{FeNO}7 have been synthesized, and characterized via 1H NMR, infrared, and electronic Accepted 30th June 2018 5 absorption spectroscopies. The ability of the FePOV–alkoxide cluster to store reducing equivalents in the DOI: 10.1039/c8sc00987b Creative Commons Attribution 3.0 Unported Licence. metalloligand for substrate coordination and activation highlights the ultility of the metal-oxide scaffold rsc.li/chemical-science as a redox reservoir. Introduction The activation of NO requires the simultaneous transfer of multiple electrons and protons to the substrate. In nature, The chemical reactivity of nitric oxide (NO) has captivated the eld similar chemical transformations of gaseous substrates (e.g. -

Lecture 6 the Crossed Aldol Reaction and Its Many Variants
Lecture 6 The Crossed Aldol Reaction and its Many Variants Objectives: By the end of this lecture you will be able to: 1. make an appropriate choice of base to completely enolise carbonyl compounds; 2. use enolates in a crossed aldol reaction; 3. recognise the aldol functional group motif, and its variants, in complex molecules; 4. use the aldol disconnection to simplify a retrosynthetic analysis; 5. use the Reformatski and Mukaiyama aldol reactions in synthesis; 6. draw arrow-pushing mechanisms for all the aldol reactions discussed in this lecture. Introduction We have seen how choosing a base (B-) whose conjugate acid (B−H) is a much poorer acid − i.e. has a much higher pKa − than the proton we wish to abstract, ensures effectively complete deprotonation of the α-C−H of a carbonyl compound to provide the corresponding enolate. By completely enolising the carbonyl group, we can suppress self-condensation aldol processes, and instead use a different electrophile such as an aldehyde to form a β-hydroxy carbonyl compound. This reaction sequence is identical in basic mechanism to the intramolecular aldol processes that we discussed in previous lectures. It is now described as a crossed aldol condensation because the electrophile is a different carbonyl compound to the one that was used to form the nucleophilic enolate. This reaction, and its many variants, provides one of the most important methods for preparing C−C bonds. Pattern Recognition The aldol reaction and its many variants are very useful reactions in synthesis. You need to be able to identify the patterns or functional group motifs where this type of bond-forming process can be used. -

Direct Synthesis of Some Significant Metal Alkoxides
SD0000032 DIRECT SYNTHESIS OF SOME SIGNIFICANT METAL ALKOXI'DE BVYU EMI1JG A THESIS SUBMITTED FOR THE DEGREE OF Mi.Sc. IN CHEMISTRY SUPERVISOR: Dr. O.Y.OMER DEPARTMENT OF CHEMISTRY FACULTY OF EDUCATION UNIVERSITY OF KHARTOUM NOVEMBER, 1998 31/28 DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document. Please be aware that all of the Missing Pages in this document were originally blank pages Dedication To my three children: Regina, Maria and Samuel CONTENTS Page Dedication i Contents ii List of Tables v List of Figures vii Acknowledgement viii Abstract (Arabic) ix Abstract (English) x CHAPTER 1.0 CHAPTER ONE - INTRODUCTION 1 2.0 CHAPTER TWO - LITERATURE REVIEW 5 2.1 Introduction to Literature Review 5 2.2 Definition of metal alkoxides 5 2.3 Metal elements and (heir chemistry 8 2.3.1 Sodium metal 8 2.3.2 Magnesium metal 12 2.3.3 Aluminium metal 16 2.3.3.1 Hydrolysis of aluminium compounds 20 2.3.4 Tin metal 21 2.4 Preparative methods and uses ofalkoxides ofNa, Mg, Al & Sn. 25 2.4.1 Sodium alkoxide 25 2.4.2 Mamiesium alkoxide 26 2.4.3 Aluminium alkoxide 27 2.4.4. Tin alkoxide 30 2.5 General properties of metal alkoxides 31 2.5.1 1 lydrolysis in metal alkoxide 34 3.0 CHAPTER THREE - MATERIALS AND EXPERIMENTAL PROCEDURE 36 3.1 General procedures 36 3.1.1 Start ing material s 3 6 3.1. I.I Apparatus 36 3.1.1.2 Dry ethanol and isopropanol 36 3.1.1.3 Na, Mg, Al & Sn metals 36 3.1.2 Infrared spectra (Ir) 37 3.2 Reactions procedures 37 3.2.1 Reaction between sodium metal and absolute ethanol 37 3.2.2 Reaction of magnesium metal with absolute ethanol 37 3.2.3 Reaction of magnesium mclal with absolute ethanol using mercury (11) chloride catalyst. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

United States Patent Office Patented May 7, 1963
3,088,959 United States Patent Office Patented May 7, 1963 1. 2 or grouping of carbon atoms which is present in cyclo 3,088,959 pentadiene. This grouping is illustrated as PROCESS OF MAKENG CYCLOPENTADEENY NECKEL, NTROSYL COMPOUNDS Robert D. Feltham, Joseph F. Anzenberger, azad Jonatian T. Carrie, Pittsburgh, Pa., assignors to The Interaa tional Nickel Company, Inc., New York, N.Y., a corpo ration of Delaware No Drawing. FiRed Sept. 1, 1960, Ser. No. 53,374 The substituent groups on the cyclopentadiene moiety 6 Clains. (C. 260-439) 0. indicated as R, R2, R3, R and R5 are any one or more The present invention relates to the production of of hydrogen atoms, halogen atoms and/or organic groups nickel compounds and, more particularly, to the produc such as aliphatic groups, aromatic groups, alicyclic groups, tion of nickel nitrosyl compounds containing a group etc. The substituent groups can also bond at two posi having the cyclopentadienyl moiety. tions. Where this occurs, groups can substitute for adja Compounds such as cyclopentadienylnickel nitrosyl, 5 cent R groups, e.g., Ra and R3 and/or R4 and R5 to form methylcyclopentadienylnickel nitrosyl and other complex indene and other condensed ring structures. nitrosyl compounds containing a cyclopentadienyl-type As mentioned hereinbefore, when carrying out the proc group have been made. Such compounds have use as ess of the present invention, the reactants are reacted in gasoline additives. When such use is contemplated, it is the presence of a base. The base can advantageously be economically imperative that the compounds be produced 20 a nitrogen base or a phosphorus base or an alkoxide of a in good yield from the most readily available and inex metal having a strong hydroxide.