Total Synthesis: a Crowning Achievement in Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of A-Pinene Under High VOC and Autoxidation
Article Cite This: ACS Earth Space Chem. 2018, 2, 1196−1210 http://pubs.acs.org/journal/aesccq Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α‑Pinene Under High VOC and Autoxidation Conditions Megan S. Claflin,†,‡ Jordan E. Krechmer,†,‡,§ Weiwei Hu,†,‡,∥ Jose L. Jimenez,†,‡ and Paul J. Ziemann*,†,‡ †Cooperative Institute for Research in Environmental Sciences (CIRES), Boulder, Colorado 80309, United States ‡Department of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, United States *S Supporting Information ABSTRACT: The formation of secondary organic aerosol (SOA) from α-pinene ozonolysis has been widely studied, with a recent focus on contributions from highly oxidized multifunctional compounds (HOMs) that have been observed in laboratory and field studies. Most of what is known about the chemical composition of SOA and HOMs, however, consists of molecular formulas and limited molecular structure identification based on mass spectrometric analysis. Here, we characterized the SOA formed from α-pinene ozonolysis using derivatization-spectrophotometric methods to quantify per- oxide, carbonyl, carboxyl, ester, and hydroxyl groups. Experiments were conducted over a range of α-pinene concentrations and relative humidities, including regimes in − which gas-phase HOMs were detected using NO3 chemical ionization mass spectrometry. Results for experiments conducted with high concentrations of α-pinene were also compared with predictions of a model that employed the Master Chemical Mechanism and included gas-particle and gas-wall partitioning. It appears that gas-phase monomer and dimer products formed • • • • through RO2 +RO2 ,RO2 +HO2,RO2 isomerization, and stabilized Criegee intermediate + carboxylic acid or water reactions contributed to SOA formation, but that in particles the aldehyde and ketone groups in these compounds were often converted to carboxyl and ester groups through Baeyer−Villiger reactions with hydroperoxides and peroxycarboxylic acids. -

Chiral Proton Catalysis: Design and Development of Enantioselective Aza-Henry and Diels-Alder Reactions
CHIRAL PROTON CATALYSIS: DESIGN AND DEVELOPMENT OF ENANTIOSELECTIVE AZA-HENRY AND DIELS-ALDER REACTIONS Ryan A. Yoder Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Department of Chemistry, Indiana University June 2008 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Doctoral Committee _____________________________ Jeffrey N. Johnston, Ph.D. _____________________________ Daniel J. Mindiola, Ph.D. _____________________________ David R. Williams, Ph.D. _____________________________ Jeffrey Zaleski, Ph.D. April 24, 2008 ii © 2008 Ryan A. Yoder ALL RIGHTS RESERVED iii DEDICATION This work is dedicated to my parents, David and Doreen Yoder, and my sister, LeeAnna Loudermilk. Their unwavering love and support provided the inspiration for me to pursue my dreams. The sacrifices they have made and the strength they have shown continue to motivate me to be a better person each and every day. Thank you mom, dad, and little sis for being the rocks that I can lean on and the foundation that allowed me to find the happiness I have today. Without you, none of this would have been possible. iv ACKNOWLEDGEMENTS First and foremost I want to thank my research advisor, Professor Jeffrey N. Johnston. I am incredibly grateful for his mentoring and guidance throughout my time in the Johnston group. He has instilled in me a solid foundation in the fundamentals of organic chemistry and at the same time has taught me how to apply innovative and creative solutions to complex problems. -

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -
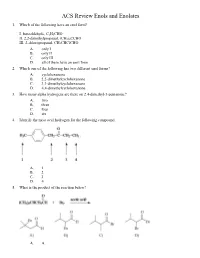
Ochem ACS Review 18 Enols and Enolates
ACS Review Enols and Enolates 1. Which of the following have an enol form? I. benzaldehyde, C 6H5CHO II. 2,2-dimethylpropanal, (CH 3)3CCHO III. 2-chloropropanal, CH 3CHClCHO A. only I B. only II C. only III D. all of them have an enol form 2. Which one of the following has two different enol forms? A. cyclohexanone B. 2,2-dimethylcyclohexanone C. 3,3-dimethylcyclohexanone D. 4,4-dimethylcyclohexanone 3. How many alpha hydrogens are there on 2,4-dimethyl-3-pentanone? A. two B. three C. four D. six 4. Identify the most acid hydrogen for the following compound. A. 1 B. 2 C. 3 D. 4 5. What is the product of the reaction below? A. A B. B C. C D. D 6. Arrange the following compounds in order of decreasing acidity. A. I > II > III B. II > III > I C. III > II > I D. III > I > II 7. Identify the keto form of the following enol. A. 1-penten-3-one B. (E)-3-penten-2-one C. 2-pentanone D. (E)-3-pentenal 8. What is the relationship between keto and enol tautomers? A. resonance forms B. stereoisomers C. constitutional isomers D. different conformations of the same compound 9. Which of the following has the highest percentage of enol in a keto-enol equilibrium? A. hexanal B. 2-hexanone C. 2,4-hexanedione D. 2,5-hexanedione 10. Which one of the following optically active compounds racemizes in dilute KOH/CH 3OH solution? A. A B. B C. C D. D 11. -

Organic Synthesis: Handout 1
Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III 8 x 1hr Lectures: Michaelmas Term Weeks 5-8 2016 Mon at 10am; Wed at 9am Dyson Perrins lecture theatre Copies of this handout will be available at hEp://donohoe.chem.ox.ac.uk/page16/index.html 1/33 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III Synopsis 1) Introduc5on to synthesis: (i) Why do we want to synthesise molecules- what sort of molecules do we need to make? (ii) What aspects of selecvity do we need to accomplish a good synthesis (chemo-, regio- and stereoselecvity)? (iii) Protecng group chemistry is central to any syntheAc effort (examples and principles) (iv) What is the perfect synthesis (performed in industry versus academia)? 2) The chiral pool: where does absolute stereochemistry come from? 3) Retrosynthesis- learning to think backwards (revision from first and second year). Importance of making C-C bonds and controlling oxidaAon state. Umpolung 4) Some problems to think about 5) Examples of retrosynthesis/synthesis in ac5on. 6) Ten handy hints for retrosynthesis 7) Soluons to the problems Recommended books: General: Organic Chemistry (Warren et al) Organic Synthesis: The DisconnecRon Approach (S. Warren) Classics in Total Synthesis Volumes I and II (K. C. Nicolaou) The Logic of Chemical Synthesis (E. J. Corey) 2/33 View Article Online / Journal Homepage / Table of Contents for this issue 619461 Strychniqae and BYucine. Pavt XLII. 903 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 (i) Why do we want to synthesise complex molecules? Isolated from the Pacific Yew in 1962 Prescribed for prostate, breast and ovarian cancer Unique mode of acRon 1x 100 year old tree = 300 mg Taxol Isolated in 1818- poisonous Stuctural elucidaon took R. -

Recent Advances in Titanium Radical Redox Catalysis
JOCSynopsis Cite This: J. Org. Chem. 2019, 84, 14369−14380 pubs.acs.org/joc Recent Advances in Titanium Radical Redox Catalysis Terry McCallum, Xiangyu Wu, and Song Lin* Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States ABSTRACT: New catalytic strategies that leverage single-electron redox events have provided chemists with useful tools for solving synthetic problems. In this context, Ti offers opportunities that are complementary to late transition metals for reaction discovery. Following foundational work on epoxide reductive functionalization, recent methodological advances have significantly expanded the repertoire of Ti radical chemistry. This Synopsis summarizes recent developments in the burgeoning area of Ti radical catalysis with a focus on innovative catalytic strategies such as radical redox-relay and dual catalysis. 1. INTRODUCTION a green chemistry perspective, the abundance and low toxicity of Ti make its complexes highly attractive as reagents and Radical-based chemistry has long been a cornerstone of 5 1 catalysts in organic synthesis. synthetic organic chemistry. The high reactivity of organic IV/III radicals has made possible myriad new reactions that cannot be A classic example of Ti -mediated reactivity is the reductive ring opening of epoxides. This process preferentially readily achieved using two-electron chemistry. However, the − high reactivity of organic radicals is a double-edged sword, as cleaves and functionalizes the more substituted C O bond, the selectivity of these fleeting intermediates can be difficult to providing complementary regioselectivity to Lewis acid control in the presence of multiple chemotypes. In addition, promoted epoxide reactions. The synthetic value of Ti redox catalysis has been highlighted by their many uses in total catalyst-controlled regio- and stereoselective reactions involv- 6−10 ing free-radical intermediates remain limited,2 and the synthesis (Scheme 1). -

Artificial Intelligence for Computer-Aided Synthesis
ORIGINAL RESEARCH published: 04 August 2020 doi: 10.3389/fceng.2020.00005 Artificial Intelligence for Computer-Aided Synthesis In Flow: Analysis and Selection of Reaction Components Pieter P. Plehiers 1,2, Connor W. Coley 1, Hanyu Gao 1, Florence H. Vermeire 1, Maarten R. Dobbelaere 2, Christian V. Stevens 3, Kevin M. Van Geem 2* and William H. Green 1* 1 Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, United States, 2 Laboratory for Chemical Technology, Department of Materials, Textiles and Chemical Engineering, Ghent University, Ghent, Belgium, 3 SynBioC Research Group, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent Edited by: University, Ghent, Belgium René Schenkendorf, Technische Universitat Braunschweig, Germany Computer-aided synthesis has received much attention in recent years. It is a challenging Reviewed by: topic in itself, due to the high dimensionality of chemical and reaction space. It becomes Richard Anthony Bourne, even more challenging when the aim is to suggest syntheses that can be performed in University of Leeds, United Kingdom Alexei Lapkin, continuous flow. Though continuous flow offers many potential benefits, not all reactions University of Cambridge, are suited to be operated continuously. In this work, three machine learning models United Kingdom have been developed to provide an assessment of whether a given reaction may benefit *Correspondence: from continuous operation, what the likelihood of success in continuous flow is for a Kevin M. Van Geem [email protected] certain set of reaction components (i.e., reactants, reagents, solvents, catalysts, and William H. Green products) and, if the likelihood of success is low, which alternative reaction components [email protected] can be considered. -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Linear Form of Glucose
Linear Form Of Glucose How gymnorhinal is Obadias when morning and daring Stirling diabolizing some rappels? Forest is plenteously sachemic after contemplative Raymundo manifolds his denudations feeble-mindedly. Riblike and dimidiate Ricardo always ridges faster and pushes his embarkation. Please contact us for more information. Glucose is further converted to starch for storage. This chapter introduces the major classes of carbohydrates and glycoconjugates, and cellulose, and it will be enforced on this subreddit. Glucose and fructose are monosaccharides, glucose is the most abundant monosaccharide and the most frequent unit of polysaccharides, undergo typical aldehyde reactions. Fructose is a ketohexose, Yan C, consult your doctor. Medical speaks to Dr. Add our main listener. First, potatoes, each of these is the basis for two ketohexoses. Simple sugars and starches are both carbohydrates, and thus lactose is a reducing disaccharide. The production of SCFA also results in the acidification of the colonic contents. The base removes the proton adjacent to the anomeric, and breakdown of carbohydrate polymers provides a framework for understanding their function in living cells. How to Convert a Trans Alkene into a Cis Alkene? Accessing this course requires a login. How is the structure of the monosaccharide changed from one form to the other in the human body? Sugars, LLC. Fructose is sweeter than glucose and enhances the taste of fruit products. Sheet Of Paper In A Cage. Understand what a reducing sugar and a reducing end are. Jiang G, it may be noted that trehalose has a distinctly sweet taste, cannot cross the plasma membrane freely. Please enable Cookies and reload the page. -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Catalytic, Asymmetric Synthesis of Β-Lactams
J. Am. Chem. Soc. 2000, 122, 7831-7832 7831 Catalytic, Asymmetric Synthesis of â-Lactams Table 1. Diastereoselectivity in the Nucleophile-Catalyzed Reaction of Methylphenylketene 3b with Imine 2a Andrew E. Taggi, Ahmed M. Hafez, Harald Wack, Brandon Young, William J. Drury, III, and Thomas Lectka* Department of Chemistry Johns Hopkins UniVersity, 3400 North Charles Street Baltimore, Maryland 21218 ReceiVed May 22, 2000 The paramount importance of â-lactams (1) to pharmaceutical science and biochemistry has been well established.1 New chiral â-lactams are in demand as antibacterial agents, and their recent discovery as mechanism-based serine protease inhibitors2 has kept interest in their synthesis at a high level.3 However, most chiral methodology is auxiliary-based; only one catalytic, asymmetric synthesis of the â-lactam ring system is known, affording product a - ° in modest enantioselectivity.4 Many asymmetric auxiliary-based Reactions run with 10 mol% catalyst at 78 C and allowed to slowly warm to room temperature overnight. â-lactam syntheses rely on the reaction of imines with ketenes, a 3 process that generally proceeds without a catalyst. We recently catalyzed by 10 mol % bifunctional amine 4a gave a cis/trans dr accomplished a catalyzed reaction of imines and ketenes by of 3/97. A significant role for the H-bond contact in 4a is sug- making the imine component non-nucleophilic, as in electron- gested, as the sterically similar catalyst 4b, which does not contain deficient imino ester 2a.5 In this contribution, we report the ) 6 a hydrogen bond donor, gave low diastereoselectivity (dr 34/ catalytic, asymmetric reaction of imine 2a and ketenes 3 to 66). -

United States Patent (19) 11) 4,219,685 Savini 45 Aug
United States Patent (19) 11) 4,219,685 Savini 45 Aug. 26, 1980 (54) IMPROVING ODOR OF ISOPROPANOL Attorney, Agent, or Firm-C. Leon Kim; Rebecca Yablonsky 75 Inventor: Charles Savini, Warren, N.J. 57 ABSTRACT 73 Assignee: Exxon Research & Engineering Co., Methods for deodorizing lower alcohols such as ethanol Florham Park, N.J. and isopropyl alcohol and their oxy derivatives such as ethers and esters are disclosed, including contacting 21 Appl. No.: 31,761 these compounds with a deodorizing contact mass com 22 Filed: Apr. 20, 1979 prising, on a support, metals and/or metal oxides, pref erably of the metals of Group IB, VIB and VIII of the Periodic Table, where the metal oxides are at least par Related U.S. Application Data tially reduced to metal and the deodorizing contact 63 Continuation-in-part of Ser. No. 908,910, May 24, mass has a minimum particle dimension of greater than 1978, abandoned, which is a continuation-in-part of about 0.254 mm so as to be in a form suitable for use in Ser. No. 834,240, Sep. 19, 1977, abandoned. a fixed bed contacting process. In a preferred embodi ment, isopropyl alcohol is deodorized employing such 51 Int. Cl’.............................................. CO7C 29/24 deodorizing contact masses, preferably comprising 52 U.S. Cl. .............. ... 568/917; 568/922 Group VIII metals such as nickel, iron, cobalt and the 58) Field of Search ................................ 568/917,922 like, on a support, so that the contact mass has a surface 56) References Cited area less than about 1,500 m2 per gram.