The Art and Science of Total Synthesis REVIEWS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules
ACCOUNT 21 Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules Shunsuke Chiba* Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore Fax +6567911961; E-mail: [email protected] Received 31 May 2012 Organic azides possess diverse chemical reactivities.4 Abstract: In this account, recent advances made on the reactions of several types of organic azides, such as vinyl azides, cyclic 2-azido Owing to their 1,3-dipole character, they undergo [3+2] alcohols, a-azido carbonyl compounds, towards the synthesis of cycloaddition with unsaturated bonds, such as those in nitrogen-containing molecules are described. alkynes and alkenes as well as carbonitriles (Scheme 1, part a).5 Organic azides can also be regarded as nitrene 1 Introduction equivalents (Scheme 1, part b).6 Accordingly, their reac- 2 Chemistry of Vinyl Azides tions with nucleophilic anions, electrophilic cations, and 2.1 Thermal [3+2]-Annulation of Vinyl Azides with 1,3-Dicar- radicals can formally provide the corresponding nitrogen bonyl Compounds 2.2 Manganese(III)-Catalyzed Formal [3+2]-Annulation with anions, cations, and radicals, respectively, forming a new 1,3-Dicarbonyl Compounds bond with the internal azido nitrogen and releasing molec- 2.3 Manganese(III)-Mediated/Catalyzed Formal [3+3]-Annu- ular nitrogen. Moreover, the generation of anions, cations, lation with Cyclopropanols and radicals at the a-position to the azido moiety can re- 2.4 Synthesis of Isoquinolines from a-Aryl-Substituted Vinyl sult in rapid denitrogenation to deliver the corresponding Azides and Internal Alkynes by Rhodium–Copper Bimetal- iminyl species, which can be used in further synthetic lic Cooperation transformations (i.e., carbon–nitrogen bond formation). -

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

Fluorescent Aminal Linked Porous Organic Polymer for Reversible Iodine Capture and Sensing Muhammad A
www.nature.com/scientificreports OPEN Fluorescent aminal linked porous organic polymer for reversible iodine capture and sensing Muhammad A. Sabri1, Mohammad H. Al‑Sayah2, Susan Sen2, Taleb H. Ibrahim1 & Oussama M. El‑Kadri2* A novel triazene-anthracene-based fuorescent aminal linked porous organic polymer (TALPOP) was prepared via metal free-Schif base polycondensation reaction of 9,10-bis-(4,6-diamino-S‑triazin‑ 2-yl)anthracene and 2-furaldehyde. The polymer has exceptional chemical and thermal stabilities and exhibit good porosity with Brunauer–Emmett–Teller surface area of 401 m2g−1. The combination of such porosity along with the highly conjugated heteroatom-rich framework enabled the polymer to exhibit exceptional iodine vapor uptake of up to 314 wt % and reversible iodine adsorption in solution. Because of the inclusion of the anthracene moieties, the TALPOP exhibited excellent 3 −1 detection sensitivity towards iodine via forescence quenching with Ksv value of 2.9 × 10 L mol . The cost efective TALPOP along with its high uptake and sensing of iodine, make it an ideal material for environmental remediation. Nuclear energy is becoming one of the most feasible alternative sources to meet the ever-increasing energy demand and minimize the emission of greenhouse gases because of its high-density energy, minimal carbon footprints, and low operation cost1–4. Despite such advantages, the potential emissions of radioactive material 129 131 3 14 85 (such as I and I, H, CO2, and Kr) from nuclear energy power plants is a major drawback of this tech- nology due to the serious environmental and health efect of these materials4,5. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -
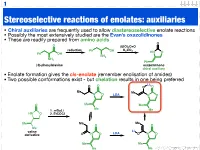
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Organic Synthesis: Handout 1
Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III 8 x 1hr Lectures: Michaelmas Term Weeks 5-8 2016 Mon at 10am; Wed at 9am Dyson Perrins lecture theatre Copies of this handout will be available at hEp://donohoe.chem.ox.ac.uk/page16/index.html 1/33 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III Synopsis 1) Introduc5on to synthesis: (i) Why do we want to synthesise molecules- what sort of molecules do we need to make? (ii) What aspects of selecvity do we need to accomplish a good synthesis (chemo-, regio- and stereoselecvity)? (iii) Protecng group chemistry is central to any syntheAc effort (examples and principles) (iv) What is the perfect synthesis (performed in industry versus academia)? 2) The chiral pool: where does absolute stereochemistry come from? 3) Retrosynthesis- learning to think backwards (revision from first and second year). Importance of making C-C bonds and controlling oxidaAon state. Umpolung 4) Some problems to think about 5) Examples of retrosynthesis/synthesis in ac5on. 6) Ten handy hints for retrosynthesis 7) Soluons to the problems Recommended books: General: Organic Chemistry (Warren et al) Organic Synthesis: The DisconnecRon Approach (S. Warren) Classics in Total Synthesis Volumes I and II (K. C. Nicolaou) The Logic of Chemical Synthesis (E. J. Corey) 2/33 View Article Online / Journal Homepage / Table of Contents for this issue 619461 Strychniqae and BYucine. Pavt XLII. 903 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 (i) Why do we want to synthesise complex molecules? Isolated from the Pacific Yew in 1962 Prescribed for prostate, breast and ovarian cancer Unique mode of acRon 1x 100 year old tree = 300 mg Taxol Isolated in 1818- poisonous Stuctural elucidaon took R. -

Acetal (POM) Chemical Compatibility Chart From
ver 31-Mar-2020 Acetal (POM) Chemical Compatibility Chart Chemical Chemical Acetaldehyde A Ammonium Acetate C Acetamide A Ammonium Bifluoride D Acetate Solvents A Ammonium Carbonate D Acetic Acid D Ammonium Caseinate D Acetic Acid, 20% C Ammonium Chloride, 10% B Acetic Acid, 80% D Ammonium Hydroxide D Acetic Acid, Glacial D Ammonium Nitrate, 10% A Acetic Anhydride D Ammonium Oxalate B Acetone A Ammonium Persulfate D Acetyl Chloride, dry D Ammonium Phosphate, Dibasic B Acetylene A Ammonium Phosphate, Monobasic B Alcohols: Amyl A Ammonium Phosphate, Tribasic B Alcohols: Benzyl A Ammonium Sulfate B Alcohols: Butyl A Ammonium Sulfite D Alcohols: Diacetone A Ammonium Thiosulfate B Alcohols: Ethyl A Amyl Acetate B Alcohols: Hexyl A Amyl Alcohol A Alcohols: Isobutyl A Amyl Chloride A Alcohols: Isopropyl A Aniline A Alcohols: Methyl A Aniline Oil D Alcohols: Octyl A Anise Oil D Alcohols: Propyl (1-Propanol) A Antifreeze D Aluminum chloride, 20% C Aqua Regia (80% HCl, 20% HNO3) D Aluminum Fluoride C Aromatic Hydrocarbons A Aluminum Hydroxide A Arsenic Acid D Aluminum Nitrate B Asphalt B Aluminum Potassium Sulfate, 10% C Barium Carbonate A Aluminum Potassium Sulfate, 100% C Barium Chloride A Aluminum Sulfate, 10% B Barium Cyanide B Alums C Barium Hydroxide D Amines D Barium Nitrate B Ammonia, 10% (Ammonium Hydroxide) C Barium Sulfate B Ammonia, 10% D Barium Sulfide A Ammonia, anhydrous D Bay Oil D Ammonia, liquid D Beer A Ammonia Nitrate C Beet Sugar Liquids B Key to General Chemical Resistance – All data is based on ambient or room temperature conditions, about 64°F (18°C) to 73°F (23°C) A = Excellent C = Fair - Moderate Effect, not recommended B= Good - Minor Effect, slight corrosion or discoloration D = Severe Effect, not recommended for ANY use It is the sole responsibility of the system designer and user to select products suitable for their specific application requirements and to ensure proper installation, operation, and maintenance of these products. -
![INDOLES from 2-METHYLNITROBENZENES by CONDENSATION with FORMAMIDE ACETALS FOLLOWED by REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(Phenylmethoxy)-]](https://docslib.b-cdn.net/cover/2671/indoles-from-2-methylnitrobenzenes-by-condensation-with-formamide-acetals-followed-by-reduction-4-benzyloxyindole-1h-indole-4-phenylmethoxy-222671.webp)
INDOLES from 2-METHYLNITROBENZENES by CONDENSATION with FORMAMIDE ACETALS FOLLOWED by REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(Phenylmethoxy)-]
DOI:10.15227/orgsyn.063.0214 Organic Syntheses, Coll. Vol. 7, p.34 (1990); Vol. 63, p.214 (1985). INDOLES FROM 2-METHYLNITROBENZENES BY CONDENSATION WITH FORMAMIDE ACETALS FOLLOWED BY REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(phenylmethoxy)-] Submitted by Andrew D. Batcho1 and Willy Leimgruber2. Checked by David J. Wustrow and Andrew S. Kende. 1. Procedure A. 6-Benzyloxy-2-nitrotoluene. A stirred mixture of 124.7 g (0.81 mol) of 2-methyl-3-nitrophenol (Note 1), 113.2 g (0.90 mol) of benzyl chloride, 112.2 g (0.81 mol) of anhydrous potassium carbonate, and 800 mL of dimethylformamide (DMF) is heated at 90°C for 3 hr. Most of the DMF is removed on a rotary evaporator (20 mm) and the oily residue is poured into 400 mL of 1 N sodium hydroxide and extracted with ether (3 × 800 mL). The combined extracts are dried (Na2SO4), filtered, and evaporated to give 203.5 g of yellowish solid. Recrystallization from 1 L of methanol cooled to 0°C affords 177.6 (90%) of 6-benzyloxy-2-nitrotoluene as pale-yellow crystals, mp 61–63°C3 (Note 2). B. (E)-6-Benzyloxy-2-nitro-β-pyrrolidinostyrene. To a solution of 175.4 g (0.72 mol) of 6- benzyloxy-2-nitrotoluene in 400 mL of DMF are added 102.5 g (0.84 mol) of N,N-dimethylformamide dimethyl acetal (Note 3) and 59.8 g (0.84 mol) of pyrrolidine. The solution is heated at reflux (110°C) for 3 hr (Note 4) under nitrogen and allowed to cool to room temperature. -

Chapter 1 Tropone and Tropolone
School of Molecular and Life Sciences New Routes to Troponoid Natural Products Jason Matthew Wells This thesis is presented for the Degree of Doctor of Philosophy of Curtin University November 2018 Declaration To the best of my knowledge and belief this thesis contains no material previously pub- lished by any other person except where due acknowledgement has been made. This thesis contains no material which has been accepted for the award of any other degree or diploma in any other university. Signature: Date: i Abstract Malaria is an infectious disease found in humans and other animals, it is caused by a single-cell parasite of the Plasmodium genus with many different substrains. Of these, P. falciparum is the most deadly to humans causing the majority of deaths. Although research into the area of antimalarial compounds is wide spread, few have been devel- oped with new structural features. Cordytropolone 37 is a natural product isolated in 2001 from the insect pathogenic fungus Cordyceps sp. BCC 1681 and has been shown to have antimalarial activity against P. falciparum. It has a structure unrelated to antimalarial com- pounds currently used in therapy. It does not contain a peroxide bridge as with artemisinin 25 or quinoline rings as with chloroquine 22. This unique structure indicates that it could possibly interact with the malaria parasite in a fashion unlike current treatments. In order for cordytropolone to be further developed as a potential treatment, it must first be synthe- sised in a laboratory environment. This study attempts to develop the first total synthesis of cordytropolone. H HO O N O O N O N H H H O O Cl HO O 22 25 37 Figure 0.0.1: Cordytropolone 37 has a unique structure compared to the current common malaria treatments The first method investigated towards the total synthesis of cordytropolone involved an intramolecular Buchner ring expansion. -

Synthesis and Structural Studies of a New Class of Quaternary Ammonium
Rivera et al. Chemistry Central Journal 2011, 5:55 http://journal.chemistrycentral.com/content/5/1/55 RESEARCHARTICLE Open Access Synthesis and structural studies of a new class of quaternary ammonium salts, which are derivatives of cage adamanzane type aminal 1, 3, 6, 8-tetraazatricyclo[4.3.1.13,8]undecane (TATU) Augusto Rivera1*, John Sadat-Bernal1, Jaime Ríos-Motta1, Michal Dušek2 and Lukáš Palatinus2 Abstract Background: Novel mono N-alkyl quaternary ammonium salts (3a-f) were prepared using the Menschutkin reaction from the cage adamanzane type aminal 1,3,6,8-tetraazatricyclo[4.3.1.13,8]undecane (TATU) and alkyl iodides, such as methyl, ethyl, propyl, butyl, pentyl and hexyl iodide (2a-f), in dry acetonitrile at room temperature. Results: The structures of these new quaternary ammonium salts were established using various spectral and electrospray ionization mass spectrometry (ESI-MS) analyses. Compound (3b) was also analyzed using X-ray crystallography. Conclusion: It was noted that alkyl chain length did not significantly affect the reaction because all employed alkyl iodide electrophiles reacted in a similar fashion with the aminal 1 to produce the corresponding mono N- quaternary ammonium salts, which were characterized by spectroscopic and analytical techniques. Background amines with haloalkyls [7]. We found that no reaction Cage aminals of the adamanzane type are tricyclic ter- occurred when N-alkylation was attempted using alkyl tiary tetraamines, which can act as bases or as nucleo- bromides and chlorides. Compound 1 reacts with alkyl philes. The main subject of research in our laboratory iodides in dry acetonitrile at room temperature to pro- (Universidad Nacional, Bogotá) is the reactivity of these duce mono N-alkyl ammonium quaternary salts (3a-f) polyamine bases toward nucleophiles and electrophiles. -

Chemical Synthesis of Carbon-14 Labeled Ricinine and Biosynthesis
1.2 PROC. OF 'nJE OKLA. ACAD. OF SCI. FOR 19M Chemical Synthesis of Carbon-14 Labeled Ridnine and Biosynthesis of Ricinine in Ricinus communis L I IL s. YANG, B. TRIPLE'rJ', IL S. )[LOS and G. B. WALLER Oldahoma State Umvenity, Acricultural Experiment station, Stillwater Ricinine (Fig. 1, tormula V; 1,2-dihydro-4-methoxy-1-methyl-2-oxo ntcotinonttrUe) is a mildly toxic alkaloid produced by the castor plant Bkiflu.t commuflu L. Studies on the biosynthesis of ricinine have been in progreu in our laboratory for several years (Waller and Henderson, 1961; Hadwiger et a!., 1963; Yang and Waller, 1965). Recently Waller et at (1Na) demoll8trated that 7~% to 90% of ricinine-'H and ricinine-8-t 'C wu degraded by the caator plant. This demonstration of metabolic acti vity servea to refute the earlier concepts that regarded alkaloids as by products of a number of irreversible and useless reactions associated with nitrogen metabolism (Pictet and Court, 1907; Cromwell, 1937; Vickery, 1941 ). To enable us to further stUdy the degradation of ricinine by the cutor plant, alkaloid labeled with carbon-14 in the pyridine ring which poueues a high specific activity is required. This report provides detailed information on the micro-scale synthesis of ricinine-3,~14C. The chemical synthesis of ricinine was initiated in the early part of this century by several workers in their attempts to prove the structure of the akaIoid. Spilth and Koller (1923) synthesized ricinine by the oxidation of 4-chloroquinoline via the intermediates 4-chloro-2-aminoquin oline-3-carboxylic acid and 2,4-dichlorontcoUnonitrile. -

Safe Handling of Sodium Azide (SAZ)
Safe Handling of Sodium Azide (SAZ) 1,2 Sodium azide (SAZ, CAS# 26628-22-8) is a white crystalline solid [molecular formula of (NaN3)] used in organic synthesis and also as a well-known preservative at low concentrations in molecular biology reagents. Azide chemistry3,4 offers an effective means to synthesize a range of nitrogen-containing compounds with a wide variety of functional groups. But SAZ poses some significant risks. It is highly toxic and can react to form potentially explosive compounds. Azide reagents and intermediates react with some metals5, strong acids, and certain chlorinated solvents6 and this needs to be considered when using SAZ or when developing routes that involve azide-containing intermediates. Health Hazards & Physical Hazards of SAZ Hazard Statements Fatal if swallowed or in contact with skin. Very toxic to aquatic life with long lasting effects. Health Hazards: SAZ is highly toxic when ingested orally or absorbed through the skin. Azides form strong complexes with hemoglobin, and consequently block oxygen transport in the blood. They are more harmful to the heart and the brain than to other organs, because the heart and the brain use a lot of oxygen. Symptoms of exposure include headache, dizziness, nausea and vomiting, rapid breathing and heart rate, and skin burns and blisters (direct skin contact) and in the case of serious overexposure, convulsions and death. It will also react with acids to form hydrazoic acid (HN3). Unlike sodium azide, which is a crystalline solid, hydrazoic acid is a low-boiling, volatile, liquid. Hydrazoic acid is also highly toxic and its volatility makes it more readily inhaled causing lung irritation and potentially bronchitis and lung edema.