Ketone and Vinyl Grignard Reagents: C- Versus O-Alkylation—Part
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Elimination Reactions E1, E2, E1cb and Ei
Elimination Reactions Dr. H. Ghosh Surendranath College, Kol-9 ________________________________________________ E1, E2, E1cB and Ei (pyrolytic syn eliminations); formation of alkenes and alkynes; mechanisms (with evidence), reactivity, regioselectivity (Saytzeff/Hofmann) and stereoselectivity; comparison between substitution and elimination. Substitution Reactions Elimination Reactions Elimination happens when the nucleophile attacks hydrogen instead of carbon Strong Base favor Elimination Bulky Nucleophile/Base favor Elimination High Temperature favors Elimination We know- This equation says that a reaction in which ΔS is positive is more thermodynamically favorable at higher temperature. Eliminations should therefore be favoured at high temperature Keep in Mind---- Mechanism Classification E1 Mechanism- Elimination Unimolecular E1 describes an elimination reaction (E) in which the rate-determining step is unimolecular (1) and does not involve the base. The leaving group leaves in this step, and the proton is removed in a separate second step E2 Mechanism- Elimination Bimolecular E2 describes an elimination (E) that has a bimolecular (2) rate-determining step that must involve the base. Loss of the leaving group is simultaneous with removal of the proton by the base Bulky t-butoxide—ideal for promoting E2 as it’s both bulky and a strong base (pKaH = 18). Other Organic Base used in Elimination Reaction These two bases are amidines—delocalization of one nitrogen’s lone pair on to the other, and the resulting stabilization of the protonated amidinium ion, E1 can occur only with substrates that can ionize to give relatively stable carbocations—tertiary, allylic or benzylic alkyl halides, for example. E1-Elimination Reaction not possible here The role of the leaving group Since the leaving group is involved in the rate-determining step of both E1 and E2, in general, any good leaving group will lead to a fast elimination. -

Proton Nmr Spectroscopy
1H NMR Spectroscopy (#1c) The technique of 1H NMR spectroscopy is central to organic chemistry and other fields involving analysis of organic chemicals, such as forensics and environmental science. It is based on the same principle as magnetic resonance imaging (MRI). This laboratory exercise reviews the principles of interpreting 1H NMR spectra that you should be learning right now in Chemistry 302. There are four questions you should ask when you are trying to interpret an NMR spectrum. Each of these will be discussed in detail. The Four Questions to Ask While Interpreting Spectra 1. How many different environments are there? The number of peaks or resonances (signals) in the spectrum indicates the number of nonequivalent protons in a molecule. Chemically equivalent protons (magnetically equivalent protons) give the same signal in the NMR whereas nonequivalent protons give different signals. 1 For example, the compounds CH3CH3 and BrCH2CH2Br all have one peak in their H NMR spectra because all of the protons in each molecule are equivalent. The compound below, 1,2- dibromo-2-methylpropane, has two peaks: one at 1.87 ppm (the equivalent CH3’s) and the other at 3.86 ppm (the CH2). 1.87 1.87 ppm CH 3 3.86 ppm Br Br CH3 1.87 ppm 3.86 10 9 8 7 6 5 4 3 2 1 0 2. How many 1H are in each environment? The relative intensities of the signals indicate the numbers of protons that are responsible for individual signals. The area under each peak is measured in the form of an integral line. -
Ch.6 Alkenes: Structure and Reactivity Alkene = Olefin
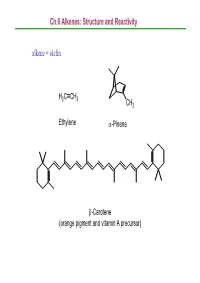
Ch.6 Alkenes: Structure and Reactivity alkene = olefin H2CCH2 CH3 Ethylene α-Pinene β-Carotene (orange pigment and vitamin A precursor) Ch.6 Alkenes: Structure and Reactivity 6.1 Industrial Preparation and Use of Alkenes Compounds derived industrially from ethylene CH3CH2OH Ethanol CH3CHO Acetaldehyde CH3COOH Acetic acid HOCH2CH2OH Ethylene glycol ClCH2CH2Cl Ethylene dichloride H C=CHCl Vinyl chloride H2CCH2 2 O Ethylene oxide Ethylene (26 million tons / yr) O Vinyl acetate O Polyethylene Ch.6 Alkenes: Structure and Reactivity Compounds derived industrially from propylene OH Isopropyl alcohol H3CCH3 O Propylene oxide CH3 H3CCH CH2 Propylene Cumene (14 million tons / yr) CH3 CH3 Polypropylene Ch.6 Alkenes: Structure and Reactivity • Ethylene, propylene, and butene are synthesized industrially by thermal cracking of natural gas (C1-C4 alkanes) and straight-run gasoline (C4-C8 alkanes). 850-900oC CH (CH ) CH H + CH + H C=CH + CH CH=CH 3 2 n 3 steam 2 4 2 2 3 2 + CH3CH2CH=CH2 - the exact processes are complex; involve radical process H 900oC CH3CH2 CH2CH3 22H2CCH H2C=CH2 +H2 Ch.6 Alkenes: Structure and Reactivity • Thermal cracking is an example of a reaction whose energetics are dominated by entropy (∆So) rather than enthalpy (∆Ho) in the free-energy equation (∆Go = ∆Ho -T∆So) . ; C-C bond cleavage (positive ∆Ho) ; high T and increased number of molecules → larger T∆So Ch.6 Alkenes: Structure and Reactivity 6.2 Calculating Degree of Unsaturation unsaturated: formula of alkene CnH2n ; formula of alkane CnH2n+2 in general, each ring or double -

Ganic Compounds
6-1 SECTION 6 NOMENCLATURE AND STRUCTURE OF ORGANIC COMPOUNDS Many organic compounds have common names which have arisen historically, or have been given to them when the compound has been isolated from a natural product or first synthesised. As there are so many organic compounds chemists have developed rules for naming a compound systematically, so that it structure can be deduced from its name. This section introduces this systematic nomenclature, and the ways the structure of organic compounds can be depicted more simply than by full Lewis structures. The language is based on Latin, Greek and German in addition to English, so a classical education is beneficial for chemists! Greek and Latin prefixes play an important role in nomenclature: Greek Latin ½ hemi semi 1 mono uni 1½ sesqui 2 di bi 3 tri ter 4 tetra quadri 5 penta quinque 6 hexa sexi 7 hepta septi 8 octa octo 9 ennea nona 10 deca deci Organic compounds: Compounds containing the element carbon [e.g. methane, butanol]. (CO, CO2 and carbonates are classified as inorganic.) See page 1-4. Special characteristics of many organic compounds are chains or rings of carbon atoms bonded together, which provides the basis for naming, and the presence of many carbon- hydrogen bonds. The valency of carbon in organic compounds is 4. Hydrocarbons: Compounds containing only the elements C and H. Straight chain hydrocarbons are named according to the number of carbon atoms: CH4, methane; C2H6 or H3C-CH3, ethane; C3H8 or H3C-CH2-CH3, propane; C4H10 or H3C-CH2- CH2-CH3, butane; C5H12 or CH3CH2CH2CH2CH3, pentane; C6H14 or CH3(CH2)4CH3, hexane; C7H16, heptane; C8H18, octane; C9H20, nonane; C10H22, CH3(CH2)8CH3, decane. -

Surface Chemistry Changes of Weathered HDPE/Wood-Flour
Polymer Degradation and Stability 86 (2004) 1–9 www.elsevier.com/locate/polydegstab Surface chemistry changes of weathered HDPE/wood-flour composites studied by XPS and FTIR spectroscopy* Nicole M. Starka,), Laurent M. Matuanab aU.S. Department of Agriculture, Forest Service, Forest Products Laboratory, One Gifford Pinchot Drive, Madison, WI 53726-2398, United States bDepartment of Forestry, Michigan State University, East Lansing, MI 48824-1222, United States Received 7 August 2003; received in revised form 3 November 2003; accepted 4 November 2003 Abstract The use of wood-derived fillers by the thermoplastic industry has been growing, fueled in part by the use of wood-fiber– thermoplastic composites by the construction industry. As a result, the durability of wood-fiber–thermoplastic composites after ultraviolet exposure has become a concern. Samples of 100% high-density polyethylene (HDPE) and HDPE filled with 50% wood- flour (WF) were weathered in a xenon arc-type accelerated weathering apparatus for 2000 h. Changes in surface chemistry were studied using spectroscopic techniques. X-ray photoelectron spectroscopy (XPS) was used to verify the occurrence of surface oxidation. Fourier transform infrared (FTIR) spectroscopy was used to monitor the development of degradation products, such as carbonyl groups and vinyl groups, and to determine changes in HDPE crystallinity. The results indicate that surface oxidation occurred immediately after exposure for both the neat HDPE and WF/HDPE composites; the surface of the WF/HDPE composites was oxidized to a greater extent than that of the neat HDPE. This suggests that the addition of WF to the HDPE matrix results in more weather-related damage. -

Electrochemistry and Photoredox Catalysis: a Comparative Evaluation in Organic Synthesis
molecules Review Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis Rik H. Verschueren and Wim M. De Borggraeve * Department of Chemistry, Molecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, box 2404, 3001 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-7693 Received: 30 March 2019; Accepted: 23 May 2019; Published: 5 June 2019 Abstract: This review provides an overview of synthetic transformations that have been performed by both electro- and photoredox catalysis. Both toolboxes are evaluated and compared in their ability to enable said transformations. Analogies and distinctions are formulated to obtain a better understanding in both research areas. This knowledge can be used to conceptualize new methodological strategies for either of both approaches starting from the other. It was attempted to extract key components that can be used as guidelines to refine, complement and innovate these two disciplines of organic synthesis. Keywords: electrosynthesis; electrocatalysis; photocatalysis; photochemistry; electron transfer; redox catalysis; radical chemistry; organic synthesis; green chemistry 1. Introduction Both electrochemistry as well as photoredox catalysis have gone through a recent renaissance, bringing forth a whole range of both improved and new transformations previously thought impossible. In their growth, inspiration was found in older established radical chemistry, as well as from cross-pollination between the two toolboxes. In scientific discussion, photoredox catalysis and electrochemistry are often mentioned alongside each other. Nonetheless, no review has attempted a comparative evaluation of both fields in organic synthesis. Both research areas use electrons as reagents to generate open-shell radical intermediates. Because of the similar modes of action, many transformations have been translated from electrochemical to photoredox methodology and vice versa. -

Novel Poly(Vinyl Alcohol)-Based Column Coating for Capillary Electrophoresis of Proteins
Biochemical Engineering Journal 53 (2010) 137–142 Contents lists available at ScienceDirect Biochemical Engineering Journal journal homepage: www.elsevier.com/locate/bej Novel poly(vinyl alcohol)-based column coating for capillary electrophoresis of proteins Liang Xu, Xiao-Yan Dong, Yan Sun ∗ Department of Biochemical Engineering and Key Laboratory of Systems Bioengineering of the Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China article info abstract Article history: A novel and simple method for the preparation of chemically bonded poly(vinyl alcohol) (PVA) coat- Received 30 June 2010 ing to silica capillary inner wall was developed, and the PVA-coated capillary columns were employed Received in revised form for capillary electrophoresis (CE). The coating procedure included pretreatment of the capillary inner 30 September 2010 wall, silanization, aldehyde group functionalization and PVA immobilization. Electroosmotic flow of the Accepted 6 October 2010 coated capillary was almost suppressed over a wide pH range (pH 3–10). High-efficiency separations of cationic proteins (including cytochrome c, lysozyme, ␣-chymotrypsinogen A) at pH 3.0–5.0 and of anionic proteins (including myoglobin and trypsin inhibitor) at pH 10.0 were achieved with the PVA-coated cap- Keywords: Protein illary. Moreover, a “dual-opposite-injection” approach was adopted for simultaneous separations of both Separation cationic and anionic proteins at neutral pH with the prepared column. In this CE mode, positively charged Bioprocess Monitoring proteins migrated from one end of the column to the detector while negatively charged proteins from Adsorption the other end to the detection window. Good run-to-run repeatability was obtained in all of the protein Capillary electrophoresis CE separations performed in this work. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

Organic Chemistry-I (Nature of Bonding and Stereochemistry
Subject Chemistry Paper No and Title 1; Organic Chemistry-I (Nature of bonding and Stereochemistry Module No and 25; Regioselectivity Title Module Tag CHE_P1_M25 CHEMISTRY Paper No. 1: Organic Chemistry-I (Nature of bonding and Stereochemistry Module no. 25: Regioselectiveity TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 2.1 Different Examples of Regioselective Reactions 2.1.1 Regioselectivity in Addition Reactions 2.1.2 Addition of HBr to Alkenes 2.1.3 Hydroboration Reaction and Addition of Hydrogen and Bromine to Alkenes 2.1.4 Baeyer Villiger Oxidation 2.1.5 Birch Reduction 2.1.6 Electrophilic Aromatic Substitution 2.1.7 Diels Alder Reaction 2.1.8 Friedal Crafts Reaction 2.1.9 Regioselectivity of Elimination 2.2 How to Determine the Regioselectivity? 3. Summary CHEMISTRY Paper No. 1: Organic Chemistry-I (Nature of bonding and Stereochemistry Module no. 25: Regioselectiveity 1. Learning Outcomes After studying this module, you shall be able to Know what is regioselectivity. Understand the basic difference between regioselectivity and chemoselectivity. Identify the regioselectivity in various reactions. Calculate the regioselectivity for a particular reaction. 2. Introduction 2. What is Regioselectivity? Regioselectivity is known as the preference chemical bond breaking or making in one direction of over all other possible directions. It is certainly applicable to positions which many reagents affect during the course of the reaction. An example to consider is which proton a strong base will abstract from an organic molecule, or where on a substituted benzene ring a further substituent will add. If there occurs a preponderance over which regioisomer will be formed, then that reaction can be termed as regioselective. -

Alkynes: Molecular and Structural Formulas
Alkynes: Molecular and Structural Formulas The alkynes comprise a series of carbon- and hydrogen-based compounds that contain at least one triple bond. This group of compounds is a homologous series with the general molecular formula of C n H2 n--2 , where n equals any integer greater than one. The simplest alkyne, ethyne (also known as acetylene), has two carbon atoms and the molecular formula of C2H2. The structural formula for ethyne is In longer alkyne chains, the additional carbon atoms are attached to each other by single covalent bonds. Each carbon atom is also attached to sufficient hydrogen atoms to produce a total of four single covalent bonds about itself. In alkynes of four or more carbon atoms, the triple bond can be located in different positions along the chain, leading to the formation of structural isomers. For example, the alkyne of molecular formula C4H6 has two isomers, Although alkynes possess restricted rotation due to the triple bond, they do not have stereoisomers like the alkenes because the bonding in a carbon-carbon triple bond is sp hybridized. In sp hybridization, the maximum separation between the hybridized orbitals is 180°, so the molecule is linear. Thus, the substituents on triple-bonded carbons are positioned in a straight line, and stereoisomers are impossible Alkynes: Nomenclature Although some common alkyne names, such as acetylene, are still found in many textbooks, the International Union of Pure and Applied Chemistry (IUPAC) nomenclature is required for journal articles. The rules for alkynes in this system are identical with those for alkenes, except for the ending. -
![[4 + 2] Annulation and Enyne Cross Metathesis](https://docslib.b-cdn.net/cover/2429/4-2-annulation-and-enyne-cross-metathesis-1192429.webp)
[4 + 2] Annulation and Enyne Cross Metathesis
Communication pubs.acs.org/JACS Gold-Catalyzed Intermolecular Reactions of Propiolic Acids with Alkenes: [4 + 2] Annulation and Enyne Cross Metathesis † † † ‡ ‡ ‡ Hyun-Suk Yeom, Jaeyoung Koo, Hyun-Sub Park, Yi Wang, Yong Liang, Zhi-Xiang Yu,*, † and Seunghoon Shin*, † Department of Chemistry and Research Institute for Natural Sciences, Hanyang University, Seoul 133-791, Korea ‡ Beijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry, Peking University, Beijing 100871, China *S Supporting Information Scheme 1. Propiolic Acid as a Functional Equivalent of 1,4- ABSTRACT: A gold-catalyzed intermolecular reaction of C,O-Dipole or Biscarbene propiolic acids with alkenes led to a [4 + 2] annulation or enyne cross metathesis. The [4 + 2] annulation proceeds with net cis-addition with respect to alkenes and provides an expedient route to α,β-unsaturated δ-lactones, for which preliminary asymmetric reactions were also demonstrated. For 1,2-disubstituted alkenes, unprecedented enyne cross metathesis occurred to give 1,3-dienes in a completely stereospecific fashion. DFT calculations and experiments indicated that the cyclobutene derivatives are not viable We commenced our study using propiolic acid (4a) and intermediates and that the steric interactions during 10 concerted σ-bond rearrangements are responsible for the styrene derivatives as substrates. After extensive optimization, observed unique stereospecificity. we found that treating styrene 3a with 4a -
![Regioselectivity in the [2 + 2] Cyclo-Addition Reaction of Triplet Carbonyl Compounds to Substituted Alkenes](https://docslib.b-cdn.net/cover/2491/regioselectivity-in-the-2-2-cyclo-addition-reaction-of-triplet-carbonyl-compounds-to-substituted-alkenes-1292491.webp)
Regioselectivity in the [2 + 2] Cyclo-Addition Reaction of Triplet Carbonyl Compounds to Substituted Alkenes
J. Chem. Sci., Vol. 117, No. 5, September 2005, pp. 561–571. © Indian Academy of Sciences. Regioselectivity in the [2 + 2] cyclo-addition reaction of triplet carbonyl compounds to substituted alkenes (Paterno–Büchi reaction): A spin-polarized conceptual DFT approach B PINTÉR,1 F DE PROFT,2 T VESZPRÉMI1 and P GEERLINGS2,* 1Inorganic Chemistry Department, Budapest University of Technology and Economics (BUTE), Szent Gellért tér 4, H-1521, Budapest, Hungary 2Eenheid Algemene Chemie (ALGC), Vrije Universiteit Brussel (VUB), Faculteit, Wetenschappen, Pleinlaan 2, 1050 Brussels, Belgium e-mail: [email protected] Abstract. Regioselectivity of the photochemical [2 + 2] cyclo-addition of triplet carbonyl compounds with a series of ground state electron-rich and electron-poor alkenes, the Paterno–Büchi reaction, is studied. Activation barriers for the first step of the triplet reaction are computed in the case of the O-attack. Next, the observed regioselectivity is explained using a series of DFT-based reactivity indices. In the first step, we use the local softness and the local HSAB principle within a softness matching approach, and explain the relative activation barriers of the addition step. In the final step, the regioselectivity is assessed within the framework of spin-polarized conceptual density functional theory, considering response functions of the system’s external potential v, number of electrons N and spin number Ns, being the difference between the number of a and b electrons in the spin-polarized system. Although the concept of local spin philicity, in- troduced recently within this theory, appears less suited to predict the regioselectivity in this reaction, the correct regioselectivity emerges from considering an interaction between the largest values of the generalized Fukui functions fss on both interacting molecules.