Chapter 2. Synthesis of Garner's Aldehyde by Asymmetric
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide
Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry at the Massachusetts Institute of Technology February, 1999 @1999 Jinhua Song All rights Reserved The author hereby grants MIT permissions to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part. Signature of Author: Department of Chemistry September 25, 1998 Certified by: Professor Satoru Masamune A. C. Cope Professor of Chemistry Thesis Supervisor Accepted by:, ProfessotDietmar Seyferth, Chairman Departmental Committee on Graduate Students MASSACHUSETTS INSTITUTE OF TECHNOLOGY LrL J This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Timothy M. Swager Chairman Professor Satoru Masamune Thesis Supervisor Professor Rick L. Danheiser , 2 Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry on September 25, 1998, in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry Abstract Presented are the stereoselective syntheses of the A (C18-C28), B (C14-C17), C (C6-C13), D (Cl-C5), C'D' (C1-C13) fragments and the efficient coupling of B and C'D' fragments of the marine natural product miyakolide, a 24-membered polyketide macrolide which exhibits anti-cancer activity. Fragment A was synthesized from the chiral aldehyde 4-4 through the successful application of the newly developed boron mediated anti-selective aldol methodology using the chiral ester 3-4. -

Synthesis of the Methyl Ester Of
Master’s Thesis 2019 60 ECTS Faculty of Chemistry, Biotechnology and Food Science Synthesis of the Methyl Ester of MaR2n-3 DPA Jeanne Sønderskov Rasmussen Master’s degree in Chemistry and Biotechnology II Acknowledgements The master’s degree was performed as collaboration between the Faculty of Chemistry, Biotechnology and Food Science at the Norwegian University of Life Sciences and the Department of Pharmacy, University of Oslo. The practical work as part of this thesis was conducted in the LIPCHEM group at the Section of Pharmaceutical Chemistry. First, I would like to thank my two supervisors, Professor Trond Vidar Hansen and Professor Yngve Stenstrøm for excellent guidance, encouragement and especially, for sharing long-time experience. Also, thanks to Dr. Jørn Tungen and Associate professor Anders Vik for solid guidance and teaching, especially during the time in the laboratory. I would also like to thank the rest of the LIPCHEM group. It has been a great experience to be a part of a research group and witness the impressive work that is carried out. Also, I am very grateful for sharing this challenging period with the four master students in pharmaceutical chemistry, Amalie Føreid Reinertsen, Marie Hermansen Mørk, Aina Kristin Pham and Margrethe Kristiansen. It has been nice to get to know you and follow your progress as well. Finally, an enormous thank to my lovely boyfriend, family and friends. Thanks for fantastic support and cheering through my six years of education. Blindern, May 2019 Jeanne Sønderskov Rasmussen III Abstract This master thesis presents the first synthesis of the methyl ester of the specialized pro-resolving mediator named MaR2n-3 DPA. -

The Synthesis and Applications of N-Alkenyl Aziridines
The Synthesis and Applications of N-Alkenyl Aziridines by Nicholas A. Afagh A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Nicholas A. Afagh 2010 The Synthesis and Applications of N-Alkenyl Aziridines Nicholas A. Afagh Master of Science Department of Chemistry University of Toronto 2010 Abstract N-alkenyl aziridines are a unique class of molecules that do not behave as typical enamines as a result of the inability of the nitrogen atom lone-pair of electrons to delocalize. The attenuated nucleophilicity of these enamines presents opportunities for the selective functionalization and reactivity not available to classical enamines. An operationally simple and mild copper-mediated coupling has been developed that facilitates the preparation of a broad range of N-alkenyl aziridines not available through existing methods. The preparation and reactivity of highly- functionalized N-alkenyl aziridines are reported. Also reported is the application of the chemoselective amine/aldehyde/alkyne (A 3) multicomponent coupling involving amphoteric aziridine aldehydes as the aldehyde component. This coupling allows access to propargyl amines with pendent aziridine functionality. ii Acknowledgments First and foremost, I would like to thank my supervisor, Professor Andrei K. Yudin for his continuous support and encouragement over the past two years. His wealth of knowledge and profound insight into all matters chemistry made for many interesting discussions. In addition, I would like to thank all the members of the Yudin group past and present with whom I have had the distinct pleasure of working alongside and shared many late evenings. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Proton Nmr Spectroscopy
1H NMR Spectroscopy (#1c) The technique of 1H NMR spectroscopy is central to organic chemistry and other fields involving analysis of organic chemicals, such as forensics and environmental science. It is based on the same principle as magnetic resonance imaging (MRI). This laboratory exercise reviews the principles of interpreting 1H NMR spectra that you should be learning right now in Chemistry 302. There are four questions you should ask when you are trying to interpret an NMR spectrum. Each of these will be discussed in detail. The Four Questions to Ask While Interpreting Spectra 1. How many different environments are there? The number of peaks or resonances (signals) in the spectrum indicates the number of nonequivalent protons in a molecule. Chemically equivalent protons (magnetically equivalent protons) give the same signal in the NMR whereas nonequivalent protons give different signals. 1 For example, the compounds CH3CH3 and BrCH2CH2Br all have one peak in their H NMR spectra because all of the protons in each molecule are equivalent. The compound below, 1,2- dibromo-2-methylpropane, has two peaks: one at 1.87 ppm (the equivalent CH3’s) and the other at 3.86 ppm (the CH2). 1.87 1.87 ppm CH 3 3.86 ppm Br Br CH3 1.87 ppm 3.86 10 9 8 7 6 5 4 3 2 1 0 2. How many 1H are in each environment? The relative intensities of the signals indicate the numbers of protons that are responsible for individual signals. The area under each peak is measured in the form of an integral line. -

S41467-018-04983-2.Pdf
ARTICLE DOI: 10.1038/s41467-018-04983-2 OPEN Novofumigatonin biosynthesis involves a non- heme iron-dependent endoperoxide isomerase for orthoester formation Yudai Matsuda 1,2,3, Tongxuan Bai2, Christopher B.W. Phippen1, Christina S. Nødvig1, Inge Kjærbølling1, Tammi C. Vesth1, Mikael R. Andersen 1, Uffe H. Mortensen 1, Charlotte H. Gotfredsen 4, Ikuro Abe 2 & Thomas O. Larsen 1 1234567890():,; Novofumigatonin (1), isolated from the fungus Aspergillus novofumigatus, is a heavily oxy- genated meroterpenoid containing a unique orthoester moiety. Despite the wide distribution of orthoesters in nature and their biological importance, little is known about the biogenesis of orthoesters. Here we show the elucidation of the biosynthetic pathway of 1 and the identification of key enzymes for the orthoester formation by a series of CRISPR-Cas9-based gene-deletion experiments and in vivo and in vitro reconstitutions of the biosynthesis. The novofumigatonin pathway involves endoperoxy compounds as key precursors for the orthoester synthesis, in which the Fe(II)/α-ketoglutarate-dependent enzyme NvfI performs the endoperoxidation. NvfE, the enzyme catalyzing the orthoester synthesis, is an Fe(II)- dependent, but cosubstrate-free, endoperoxide isomerase, despite the fact that NvfE shares sequence homology with the known Fe(II)/α-ketoglutarate-dependent dioxygenases. NvfE thus belongs to a class of enzymes that gained an isomerase activity by losing the α- ketoglutarate-binding ability. 1 Department of Biotechnology and Biomedicine, Technical University of Denmark, Søltofts Plads, 2800 Kgs. Lyngby, Denmark. 2 Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. 3 Department of Chemistry, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong SAR, China. -

Publikationsliste Internet
Professor Dr. Alois Fürstner List of Publications 2020 F. Caló, A. Fürstner A Heteroleptic Dirhodium Catalyst for Asymmetric Cyclopropanation with -Stannyl -Diazoacetate. ‘Stereoretentive’ Stille Coupling with Formation of Chiral Quarternary Carbon Centers Angew. Chem. 2020, 132; 14004-14011, Angew. Chem. Int. Ed. 2020, 59, 13900-13907 H. Jin, A. Fürstner Modular Synthesis of Furans with up to Four Different Substituents by a trans‐Carboboration Strategy Angew. Chem. 2020, 132; 13720-13724, Angew. Chem. Int. Ed. 2020, 29, 1316-13622 Z. Meng, A. Fürstner Total Synthesis Provides Strong Evidence: Xestocyclamine A is the Enantiomer of Ingenamine J. Am. Chem. Soc. 2020, 142, 11703-11708 J. Hillenbrand, M. Leutzsch, E. Yiannakas, C. Gordon, C. Wille, N. Nöthling, C. Copéret, A. Fürstner “Canopy Catalysts” for Alkyne Metathesis: Molybdenum Alkylidyne Complexes with a Tripodal Ligand Framework J. Am. Chem. Soc.2020, 142, 11279-11294 M. Heinrich, J. Murphy, M. Ilg, A. Letort, J. Flasz, P. Philipps, A. Fürstner Chagosensine: A Riddle Wrapped in a Mystery Inside an Enigma J. Am. Chem. Soc. 2020, 142, 6409-6422 M. Buchsteiner, L. Martinez-Rodriguez, P. Jerabek, I. Pozo, M. Patzer, N. Nöthling, C. Lehmann, A. Fürstner Catalytic Asymmetric Fluorination of Copper Carbene Complexes: Preparative Advances and a Mechanistic Rationale Chem.–Eur. J. 2020, 26, 2509-2515 2019 S. Peil, A. Fürstner Mechanistic Divergence in the Hydrogenative Synthesis of Furans and Butenolides: Ruthenium Carbenes Formed by gem-Hydrogenation or via Carbophilic Activation of Alkynes Angew. Chem. 2019, 131; 18647-18652; Angew. Chem. Int. Ed. 2019, 58, 18476-18481 L. Huang, Y. Gu, A. Fürstner Iron Catalyzed Reactions of 2-Pyridone Derivatives: 1,6-Addition and Formal Ring Opening/Cross Coupling Chem. -
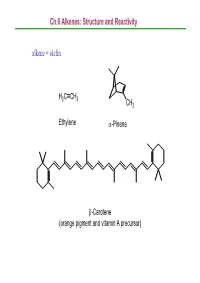
Ch.6 Alkenes: Structure and Reactivity Alkene = Olefin
Ch.6 Alkenes: Structure and Reactivity alkene = olefin H2CCH2 CH3 Ethylene α-Pinene β-Carotene (orange pigment and vitamin A precursor) Ch.6 Alkenes: Structure and Reactivity 6.1 Industrial Preparation and Use of Alkenes Compounds derived industrially from ethylene CH3CH2OH Ethanol CH3CHO Acetaldehyde CH3COOH Acetic acid HOCH2CH2OH Ethylene glycol ClCH2CH2Cl Ethylene dichloride H C=CHCl Vinyl chloride H2CCH2 2 O Ethylene oxide Ethylene (26 million tons / yr) O Vinyl acetate O Polyethylene Ch.6 Alkenes: Structure and Reactivity Compounds derived industrially from propylene OH Isopropyl alcohol H3CCH3 O Propylene oxide CH3 H3CCH CH2 Propylene Cumene (14 million tons / yr) CH3 CH3 Polypropylene Ch.6 Alkenes: Structure and Reactivity • Ethylene, propylene, and butene are synthesized industrially by thermal cracking of natural gas (C1-C4 alkanes) and straight-run gasoline (C4-C8 alkanes). 850-900oC CH (CH ) CH H + CH + H C=CH + CH CH=CH 3 2 n 3 steam 2 4 2 2 3 2 + CH3CH2CH=CH2 - the exact processes are complex; involve radical process H 900oC CH3CH2 CH2CH3 22H2CCH H2C=CH2 +H2 Ch.6 Alkenes: Structure and Reactivity • Thermal cracking is an example of a reaction whose energetics are dominated by entropy (∆So) rather than enthalpy (∆Ho) in the free-energy equation (∆Go = ∆Ho -T∆So) . ; C-C bond cleavage (positive ∆Ho) ; high T and increased number of molecules → larger T∆So Ch.6 Alkenes: Structure and Reactivity 6.2 Calculating Degree of Unsaturation unsaturated: formula of alkene CnH2n ; formula of alkane CnH2n+2 in general, each ring or double -

Properties of Silicon Hafensteiner
Baran Lab Properties of Silicon Hafensteiner Si vs. C Siliconium Ion - Si is less electronegative than C - Not believed to exist in any reaction in solution - More facile nucleophilic addition at Si center J. Y. Corey, J. Am. Chem. Soc. 1975, 97, 3237 - Pentacoordinate Si compounds have been observed Average BDE (kcal/mol) MeSiF4 NEt4 Ph3SiF2 NR4 C–C C–Si Si–Si C–F Si–F 83 76 53 116 135 - Lack of cation justified by high rate of bimolecular reactivity at Si C–O Si–O C–H Si–H Mechanism of TMS Deprotection 86 108 83 76 OTMS O Average Bond Lengths (Å) C–C C–Si C–O Si–O 1.54 1.87 1.43 1.66 Workup Si Si Silicon forms weak p-Bonds O O F F NBu4 p - C–C = 65 kcal/mol p - C–Si = 36 kcal/mol Pentavalent Silicon Baran Lab Properties of Silicon Hafensteiner Nucleophilic addition to Si b-Silicon effect and Solvolysis F RO–SiMe3 RO F–SiMe3 SiMe3 Me H H vs. Me3C H Me3C H OSiMe O Li 3 H OCOCF H OCOCF MeLi 3 3 A B Me-SiMe3 12 kA / kB = 2.4 x 10 Duhamel et al. J. Org. Chem. 1996, 61, 2232 H H SiMe Me b-Silicon Effect 3 vs. Me3C H Me3C H - Silicon stabalizes b-carbocations H OCOCF3 H OCOCF3 - Stabalization is a result of hyperconjugation 4 kA / kB = 4 x 10 SiR3 CR3 Evidence for Stepwise mechanism vs. Me3Si SiMe2Ph SiMe2Ph A B Me3Si SiMe2Ph *A is more stable than B by 38 kcal/mol * Me3Si SiMe2Ph Me3Si Jorgensen, JACS, 1986,107, 1496 Product ratios are equal from either starting material suggesting common intermediate cation Baran Lab Properties of Silicon Hafensteiner Evidence for Rapid Nucleophilic Attack Extraordinary Metallation Me SiMe3 SiMe3 Li Si t-BuLi Me SnCl4 Cl Me3Si Cl Me2Si Cl SiMe MeO OMe 3 OMe OMe Me2Si Cl vs. -

Ganic Compounds
6-1 SECTION 6 NOMENCLATURE AND STRUCTURE OF ORGANIC COMPOUNDS Many organic compounds have common names which have arisen historically, or have been given to them when the compound has been isolated from a natural product or first synthesised. As there are so many organic compounds chemists have developed rules for naming a compound systematically, so that it structure can be deduced from its name. This section introduces this systematic nomenclature, and the ways the structure of organic compounds can be depicted more simply than by full Lewis structures. The language is based on Latin, Greek and German in addition to English, so a classical education is beneficial for chemists! Greek and Latin prefixes play an important role in nomenclature: Greek Latin ½ hemi semi 1 mono uni 1½ sesqui 2 di bi 3 tri ter 4 tetra quadri 5 penta quinque 6 hexa sexi 7 hepta septi 8 octa octo 9 ennea nona 10 deca deci Organic compounds: Compounds containing the element carbon [e.g. methane, butanol]. (CO, CO2 and carbonates are classified as inorganic.) See page 1-4. Special characteristics of many organic compounds are chains or rings of carbon atoms bonded together, which provides the basis for naming, and the presence of many carbon- hydrogen bonds. The valency of carbon in organic compounds is 4. Hydrocarbons: Compounds containing only the elements C and H. Straight chain hydrocarbons are named according to the number of carbon atoms: CH4, methane; C2H6 or H3C-CH3, ethane; C3H8 or H3C-CH2-CH3, propane; C4H10 or H3C-CH2- CH2-CH3, butane; C5H12 or CH3CH2CH2CH2CH3, pentane; C6H14 or CH3(CH2)4CH3, hexane; C7H16, heptane; C8H18, octane; C9H20, nonane; C10H22, CH3(CH2)8CH3, decane. -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

Surface Chemistry Changes of Weathered HDPE/Wood-Flour
Polymer Degradation and Stability 86 (2004) 1–9 www.elsevier.com/locate/polydegstab Surface chemistry changes of weathered HDPE/wood-flour composites studied by XPS and FTIR spectroscopy* Nicole M. Starka,), Laurent M. Matuanab aU.S. Department of Agriculture, Forest Service, Forest Products Laboratory, One Gifford Pinchot Drive, Madison, WI 53726-2398, United States bDepartment of Forestry, Michigan State University, East Lansing, MI 48824-1222, United States Received 7 August 2003; received in revised form 3 November 2003; accepted 4 November 2003 Abstract The use of wood-derived fillers by the thermoplastic industry has been growing, fueled in part by the use of wood-fiber– thermoplastic composites by the construction industry. As a result, the durability of wood-fiber–thermoplastic composites after ultraviolet exposure has become a concern. Samples of 100% high-density polyethylene (HDPE) and HDPE filled with 50% wood- flour (WF) were weathered in a xenon arc-type accelerated weathering apparatus for 2000 h. Changes in surface chemistry were studied using spectroscopic techniques. X-ray photoelectron spectroscopy (XPS) was used to verify the occurrence of surface oxidation. Fourier transform infrared (FTIR) spectroscopy was used to monitor the development of degradation products, such as carbonyl groups and vinyl groups, and to determine changes in HDPE crystallinity. The results indicate that surface oxidation occurred immediately after exposure for both the neat HDPE and WF/HDPE composites; the surface of the WF/HDPE composites was oxidized to a greater extent than that of the neat HDPE. This suggests that the addition of WF to the HDPE matrix results in more weather-related damage.