Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -
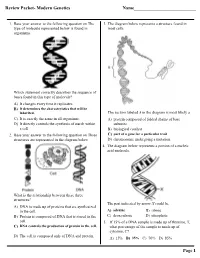
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Application of X-Ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-19-2008 Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis Zakhia Moore University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Moore, Zakhia, "Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis" (2008). University of New Orleans Theses and Dissertations. 888. https://scholarworks.uno.edu/td/888 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Chemistry by Zakhia Moore B.S., Xavier University, 2000 M.S., University of New Orleans, 2005 December, 2008 ACKNOWLEDGEMENTS Giving all glory to God for His many blessings, whom without I could not have completed this long journey. -

IAPS Act 15-17 Section Preview of the Teachers Edition.Pdf
Section Preview of the Teacher’s Edition for The Chemistry of Materials, Issues and Physical Science, 2nd Edition Activities 15-17 Suggested student responses and answer keys have been blocked out so that web-savvy students do not find this page and have access to answers. To experience a complete activity please request a sample found in the footer at lab-aids.com Families of Elements 15 4 s 0 2 n - o t si o es 50 te s -minu N O I T I N A ACTIVITY OVERVIEW V E S T I G Students compare physical and chemical properties of 13 elements and sort them into groups based on common properties. They then compare their classifications with groups—or families—of elements as defined by scientists and displayed in the Periodic Table of the Elements. KEY CONCEPTS AND PROCESS SKILLS (with correlation to NSE 5–8 Content Standards) 1. Elements have characteristic physical and chemical properties. Substances often are placed in categories or groups if they react in similar ways; metals are an example of such a group. (PhysSci: 1) 2. Scientists classify elements into families, based on their properties. (PhysSci: 1) 3. Students develop descriptions based on evidence. (Inquiry: 1) KEY VOCABULARY atom atomic mass element family (of elements) metal Periodic Table of the Elements B-39 Activity 15 • Families of Elements MATERIALS AND ADVANCE PREPARATION For the teacher aluminum, iron, copper, and carbon samples from Activity 14, “Physical and Chemical Properties of Materials” 1 Transparency 15.1, “Information on Element Cards” 8 sets of 4 Element Family Cards -

Molecular Model-Building by Computer
Molecular Model-building by Computer In which biochemists observe models of giant molecules as they are displayed on a screen by a computer and try to fold them into the shapes that they assume in nature by Cyrus Levinthal any problems of modern biology a large number of problems in biology computer to help gain such information are concerned with the detailed into which biologists have some insight about the structure of large biological M relation between biological but concerning which they cannot yet molecules. function and molecular structure. Some ask suitable questions in terms of molec- For the first half of this century the of the questions currently being asked ular structure. As they see such prob- metabolic and structural relations among will be completely answered only when lems more clearly, however, they in- the small molecules of the living cell one has an understanding of the struc- variably find an increasing need for were the principal concern of bio- ture of all the molecular components of structural information. In our laboratory chemists. The chemical reactions these a biological system and a knowledge of at the Massachusetts Institute of Tech- molecules undergo have been studied how they interact. There are, of course, nology we have recently started using a intensively. Such reactions are specifical- ly catalyzed by the large protein mole- cules called enzymes, many of which have now been purified and also studied. It is only within the past few years, however, that X-ray-diffraction techniques have made it possible to determine the molecular structure of such protein molecules. -

CORINA Classic Program Manual
CORINA Classic Generation of Three-Dimensional Molecular Models Version 4.4.0 Program Description Molecular Networks GmbH September 2021 www.mn-am.com Molecular Networks GmbH Altamira LLC Neumeyerstraße 28 470 W Broad St, Unit #5007 90411 Nuremberg Columbus, OH 43215 Germany USA mn-am.com This document is copyright © 1998-2021 by Molecular Networks GmbH Computerchemie and Altamira LLC. All rights reserved. Except as permitted under the terms of the Software Licensing Agreement of Molecular Networks GmbH Computerchemie, no part of this publication may be reproduced or distributed in any form or by any means or stored in a database retrieval system without the prior written permission of Molecular Networks GmbH or Altamira LLC. The software described in this document is furnished under a license and may be used and copied only in accordance with the terms of such license. (Document version: 4.4.0-2021-09-30) Content Content 1 Introducing CORINA Classic 1 1.1 Objective of CORINA Classic 1 1.2 CORINA Classic in Brief 1 2 Release Notes 3 2.1 CORINA (Full Version) 3 2.2 CORINA_F (Restricted FlexX Interface Version) 25 3 Getting Started with CORINA Classic 26 4 Using CORINA Classic 29 4.1 Synopsis 29 4.2 Options 29 5 Use Cases of CORINA Classic 60 6 Supported File Formats and Interfaces 64 6.1 V2000 Structure Data File (SD) and Reaction Data File (RD) 64 6.2 V3000 Structure Data File (SD) and Reaction Data File (RD) 67 6.3 SMILES Linear Notation 67 6.4 InChI file format 69 6.5 SYBYL File Formats 70 6.6 Brookhaven Protein Data Bank Format (PDB) -

Ab Initio Solution of Macromolecular Crystal Structures Without Direct Methods
Ab initio solution of macromolecular crystal structures without direct methods Airlie J. McCoya, Robert D. Oeffnera, Antoni G. Wrobelb, Juha R. M. Ojalac, Karl Tryggvasonc,d, Bernhard Lohkampe, and Randy J. Reada,1 aDepartment of Haematology, Cambridge Institute for Medical Research, University of Cambridge, Cambridge CB2 0XY, United Kingdom; bDepartment of Clinical Biochemistry, Cambridge Institute for Medical Research, University of Cambridge, Cambridge CB2 0XY, United Kingdom; cDivision of Matrix Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, 171 77 Stockholm, Sweden; dCardiovascular and Metabolic Disorders Program, Duke-NUS (National University of Singapore) Medical School, 16957 Singapore; and eDivision of Molecular Structural Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, 171 77 Stockholm, Sweden Edited by Axel T. Brunger, Stanford University, Stanford, CA, and approved February 27, 2017 (received for review January 30, 2017) The majority of macromolecular crystal structures are determined (optionally) a correction for the effect of disordered solvent using the method of molecular replacement, in which known described by the parameters fsol and Bsol: related structures are rotated and translated to provide an initial sffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi atomic model for the new structure. A theoretical understanding 2 2 Bsol 2π Δ of the signal-to-noise ratio in likelihood-based molecular replace- σA = fP 1 − fsolexp − exp − , [1a] d2 d2 ment searches has been developed to account for the influence of 4 3 model quality and completeness, as well as the resolution of the diffraction data. Here we show that, contrary to current belief, pffiffiffiffi 2π2 Δ2 molecular replacement need not be restricted to the use of models σA ≈ fPexp − . -
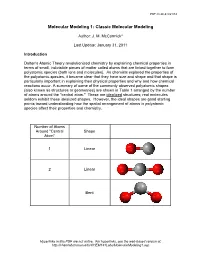
Molecular-Modeling-1.Pdf
PDF created 8/21/14 Molecular Modeling 1: Classic Molecular Modeling Author: J. M. McCormick* Last Update: January 31, 2011 Introduction Dalton's Atomic Theory revolutionized chemistry by explaining chemical properties in terms of small, indivisible pieces of matter called atoms that are linked together to form polyatomic species (both ions and molecules). As chemists explored the properties of the polyatomic species, it became clear that they have size and shape and that shape is particularly important in explaining their physical properties and why and how chemical reactions occur. A summary of some of the commonly observed polyatomic shapes (also known as structures or geometries) are shown in Table 1 arranged by the number of atoms around the "central atom." These are idealized structures; real molecules seldom exhibit these idealized shapes. However, the ideal shapes are good starting points toward understanding how the spatial arrangement of atoms in polyatomic species affect their properties and chemistry. Number of Atoms Around "Central Shape Atom" 1 Linear 2 Linear Bent Hyperlinks in this PDF are not active. For hyperlinks, see the web-based version at: http://chemlab.truman.edu/CHEM131Labs/MolecularModeling1.asp PDF created 8/21/14 3 Trigonal planar Trigonal pyramidal T-shape 4 Tetrahedral Hyperlinks in this PDF are not active. For hyperlinks, see the web-based version at: http://chemlab.truman.edu/CHEM131Labs/MolecularModeling1.asp PDF created 8/21/14 Square planar Bisphenoid (see-saw) 5 Trigonal bipyramidal Square pyramidal 6 Octahedral Hyperlinks in this PDF are not active. For hyperlinks, see the web-based version at: http://chemlab.truman.edu/CHEM131Labs/MolecularModeling1.asp PDF created 8/21/14 Table 1. -

Kitamo Molecular Models Ball-And-Stick Model Design Guide
KitaMo Molecular Models Ball-and-Stick Model Design Guide Draft Version. Issue Date 10 April 2012. WARNING: Please bear in mind that our models are not toys. Allow children to handle them only with adult supervision. Although they are built with structural integrity, they can easily be damaged by mishandling. KitaMo Molecular Models are custom-made to your specifications. This Guide is intended to help you prepare the specifications for your Ball-and-Stick Molecular Model. If your requirements are not discussed in this guide, please let us know: [email protected]. Copyright © 2012 KitaMo Molecular Models. All rights reserved. Do not quench your inspiration and your imagination; do not become the slave of your model. Vincent Van Gogh KitaMo Molecular Models are custom-built using precisely-machined parts. They are not assembled from commercially available kits, resulting in accurate structures. Ball-and-Stick models are typically built on a scale of 1 Angstrom per centimeter (10 million times actual size), a compact scale that allows for easy handling and requires little display space. The tools and techniques used to produce such miniatures may be adapted to build larger scale models. To order a custom-made molecular model, the following information are required: structure, scale, color scheme, accuracy and display preference. Structural Information about the molecule. From this the number of atoms, the number of bonds, the bond angles and the bond lengths can all be determined. Such information may be provided using atomic coordinates preferably in an electronic file format that can be read by JMol (e.g., PDB). -

Polyhedron Molecular Model
HGS POLYHEDRON MOLECULAR MODEL New Alpha Series Manual 3 rd Edition #1000α Fundamental Organic Chemistry Set #1001α General Chemistry Basic Set #1002α Organic Chemistry Introductory Set #1013α Organic Chemistry Set for Student #1003α Organic Chemistry Basic Set #1005α Organic Chemistry Standard Set Tokyo, Japan Polyhedron Molecular Model Sets: Contents ■Atom Atom Quantity Item No. Color Use α α α #1013α #1003α #1005α Name #1000 #1001 #1002 3rd Edition 2nd Edition 3rd Edition ATM-01 H white s 30 24 24 30 30 72 ATM-11 B orange sp2, dsp3 - - - - - 1 ATM-02 C(sp3) black sp3 12 12 12 12 28 46 ATM-09 C(sp2) black sp2, dsp3 - 3 - 6 12 20 ATM-03 N(sp3) blue sp3 2 6 2 1 3 5 ATM-10 N(sp2) blue sp2, dsp3 - - - 1 3 5 ATM-04 O red sp3 6 2 2 4 7 12 yellow ATM-05 Si sp3 - green - - - - 1 ATM-06 P yellow sp3 - - - - 1 2 ATM-07 S pink sp3 - - - 1 1 2 ATM-08 Cl green sp3 2 2 - 1 2 4 3 ATM-17 m(sp) gray sp, sp , - 3 1 2 2 4 d2sp3 ■Bond (1Å=2.5cm) Quantity Bond Item No. (Å) Color Use α α α #1013α #1003α #1005α Length #1000 #1001 #1002 3rd Edition 2nd Edition 3rd Edition 1.09*1 C-H BND-02 pink 30 25 25 30 30 72 1.21*2 C≡C,C=O C=C,C-O BND-04 1.40 - - - 8 16 38 green C(ar)=C(ar) BND-06 1.54 white C-C, S-O 20 20 20 24 40 72 C-P BND-07 1.80 yellow - - - 2 8 20 C-Cl, C-S BND-09 2.10 white C-I - - - - - 2 BND-10 1.33 blue C=C 12 6 6 10 12 30 (bent bond) *1) In the case of C–(#2 bond)–H, bond length = 1.09 Å. -

Chem 351: Molecular Models
MOD.1 MOLECULAR STRUCTURES AND MODELS Note: There will be no pre-laboratory quiz for this experiment. However, you should start work on the content. The laboratory period will run more like a tutorial, it’s “open book”, you can work in groups and ask your TA about any concepts you don't understand. This laboratory activity is assessed based on an online Moodle “molecular models” graded activity (30 min. time limit (including 50% technology time)) that is to be completed within 48 hrs of your in-person laboratory session. This activity is essentially the same as one we have successfully used for many years to help students with the visualization of molecules and to learn how to use their model kits. Model kits are a tool that chemists will use to visualize and explain aspects of chemical structure. They are very useful for helping students develop the ability to visualize molecules in 3 dimensions (after all, very few molecules are actually flat). Some students certainly struggle to grasp and manage this stereochemistry without the use of model kits (like the one shown below which it the type we typically recommend). Since we regard model kits as a valuable tool, and a tool a chemist might use, for many years we have allowed model kits to be used during examinations. We also know that many students don’t really know how to use model kits correctly and effectively so this activity tries to show that while also exploring some to the topics related to stereochemistry that many students often struggle with. -
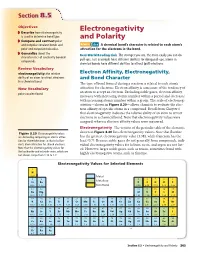
Electronegativity and Polarity 265 EN Difference Table 8.7 and Bond Character
Section 88.5.5 Objectives Electronegativity ◗ Describe how electronegativity is used to determine bond type. and Polarity ◗ Compare and contrast polar and nonpolar covalent bonds and MAIN Idea A chemical bond’s character is related to each atom’s polar and nonpolar molecules. attraction for the electrons in the bond. ◗ Generalize about the Real-World Reading Link The stronger you are, the more easily you can do characteristics of covalently bonded pull-ups. Just as people have different abilities for doing pull-ups, atoms in compounds. chemical bonds have different abilities to attract (pull) electrons. Review Vocabulary electronegativity: the relative Electron Affinity, Electronegativity, ability of an atom to attract electrons and Bond Character in a chemical bond The type of bond formed during a reaction is related to each atom’s New Vocabulary attraction for electrons. Electron affinity is a measure of the tendency of an atom to accept an electron. Excluding noble gases, electron affinity polar covalent bond increases with increasing atomic number within a period and decreases with increasing atomic number within a group. The scale of electroneg- ativities—shown in Figure 8.20—allows chemists to evaluate the elec- tron affinity of specific atoms in a compound. Recall from Chapter 6 that electronegativity indicates the relative ability of an atom to attract electrons in a chemical bond. Note that electronegativity values were assigned, whereas electron affinity values were measured. Electronegativity The version of the periodic table of the elements Figure 8.20 Electronegativity values shown in Figure 8.20 lists electronegativity values. Note that fluorine are derived by comparing an atom’s attrac- has the greatest electronegativity value (3.98), while francium has the tion for shared electrons to that of a fluo- least (0.7).