Molecular Property Diagnostic Suite for Tuberculosis (MPDSTB)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -
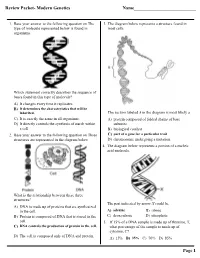
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Application of X-Ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-19-2008 Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis Zakhia Moore University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Moore, Zakhia, "Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis" (2008). University of New Orleans Theses and Dissertations. 888. https://scholarworks.uno.edu/td/888 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Chemistry by Zakhia Moore B.S., Xavier University, 2000 M.S., University of New Orleans, 2005 December, 2008 ACKNOWLEDGEMENTS Giving all glory to God for His many blessings, whom without I could not have completed this long journey. -

Open Babel Documentation Release 2.3.1
Open Babel Documentation Release 2.3.1 Geoffrey R Hutchison Chris Morley Craig James Chris Swain Hans De Winter Tim Vandermeersch Noel M O’Boyle (Ed.) December 05, 2011 Contents 1 Introduction 3 1.1 Goals of the Open Babel project ..................................... 3 1.2 Frequently Asked Questions ....................................... 4 1.3 Thanks .................................................. 7 2 Install Open Babel 9 2.1 Install a binary package ......................................... 9 2.2 Compiling Open Babel .......................................... 9 3 obabel and babel - Convert, Filter and Manipulate Chemical Data 17 3.1 Synopsis ................................................. 17 3.2 Options .................................................. 17 3.3 Examples ................................................. 19 3.4 Differences between babel and obabel .................................. 21 3.5 Format Options .............................................. 22 3.6 Append property values to the title .................................... 22 3.7 Filtering molecules from a multimolecule file .............................. 22 3.8 Substructure and similarity searching .................................. 25 3.9 Sorting molecules ............................................ 25 3.10 Remove duplicate molecules ....................................... 25 3.11 Aliases for chemical groups ....................................... 26 4 The Open Babel GUI 29 4.1 Basic operation .............................................. 29 4.2 Options ................................................. -

JRC QSAR Model Database
JRC QSAR Model Database EURL ECVAM DataBase service on ALternative Methods to animal experimentation To promote the development and uptake of alternative and advanced methods in toxicology and biomedical sciences SDF - STRUCTURE DATA FORMAT: How to create from SMILES The European Commission’s science and knowledge service Joint Research Centre Directorate F Health, Consumers & Reference Materials Chemicals Safety & Alternative Methods Unit The European Commission’s science and knowledge service Joint Research Centre EUR 28708 EN This publication is a Tutorial by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide user support. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Email: [email protected] JRC Science Hub https://ec.europa.eu/jrc JRC107492 EUR 28708 EN PDF ISBN 978-92-79-71294-4 ISSN 1831-9424 doi:10.2760/952280 Print ISBN 978-92-79-71295-1 ISSN 1018-5593 doi:10.2760/668595 Luxembourg: Publications Office of the European Union, 2017 Ispra: European Commission, 2017 © European Union, 2017 The reuse of the document is authorised, provided the source is acknowledged and the original meaning or message of the texts are not distorted. The European Commission shall not be held liable for any consequences stemming from the reuse. How to cite this document: Triebe -

IAPS Act 15-17 Section Preview of the Teachers Edition.Pdf
Section Preview of the Teacher’s Edition for The Chemistry of Materials, Issues and Physical Science, 2nd Edition Activities 15-17 Suggested student responses and answer keys have been blocked out so that web-savvy students do not find this page and have access to answers. To experience a complete activity please request a sample found in the footer at lab-aids.com Families of Elements 15 4 s 0 2 n - o t si o es 50 te s -minu N O I T I N A ACTIVITY OVERVIEW V E S T I G Students compare physical and chemical properties of 13 elements and sort them into groups based on common properties. They then compare their classifications with groups—or families—of elements as defined by scientists and displayed in the Periodic Table of the Elements. KEY CONCEPTS AND PROCESS SKILLS (with correlation to NSE 5–8 Content Standards) 1. Elements have characteristic physical and chemical properties. Substances often are placed in categories or groups if they react in similar ways; metals are an example of such a group. (PhysSci: 1) 2. Scientists classify elements into families, based on their properties. (PhysSci: 1) 3. Students develop descriptions based on evidence. (Inquiry: 1) KEY VOCABULARY atom atomic mass element family (of elements) metal Periodic Table of the Elements B-39 Activity 15 • Families of Elements MATERIALS AND ADVANCE PREPARATION For the teacher aluminum, iron, copper, and carbon samples from Activity 14, “Physical and Chemical Properties of Materials” 1 Transparency 15.1, “Information on Element Cards” 8 sets of 4 Element Family Cards -

MACSIMUS Manual
1 MACSIMUS manual benzocaine (ethylaminobenzoate) parameter_set = charmm21 HA | HA-CT-HA | HA-CT-HA | OSn.2 | Cp.7=On.5 | C6R--C6R-HA Most often used links: | | HA-C6R C6R-HA 2.2 Blend synopsis and options | | HA-C6R--C6R-NPn.5^-Hp.25 9.2 Cook synopsis and options | 9.2.5 Cook input data Hp.25 MACromolecule SIMUlation Software © Jiˇr´ıKolafa 1993{2020 MACSIMUS may be distributed under the terms of the GNU General Public Licence Credits: ray: Mark VandeWettering \reasonably intelligent raytracer" CHARMM force field (files: charmm*.par, charmm*/*.rsd) GROMOS force field (files: gromos*.par, gromos*/*.rsd) amoeba implementation by Z. Wagner moil support by J. Schofield several bug fixes by T. Trnka bug discovered by N. Parfenov Contents I Program `blend' version 2.4b 14 1 Introduction 16 1.1 Force fields...................................... 16 1.2 `blend' overview.................................... 16 1.3 Versions........................................ 17 2 Running blend 18 2.1 Environment...................................... 18 2.2 Synopsis........................................ 19 2.2.1 Global options................................. 19 2.2.2 par-options and parameter files....................... 20 2.2.3 mol-options and molecular files....................... 22 2.2.4 Extra-options ................................. 29 2.3 File extensions.................................... 34 2.4 Run-time control................................... 37 2.4.1 get data format for input........................... 37 2.4.2 Scrolling.................................... 38 2.4.3 Error handling................................ 39 2.4.4 Interrupts................................... 40 2.5 Showing molecules graphically............................ 40 2.5.1 X11 Graphics................................. 40 2.5.2 Playback output............................... 44 2.6 Energy minimization................................. 44 2.7 Missing coordinates.................................. 45 3 Force field and the parameter file 46 3.1 Structure of the parameter file........................... -

Molecular Model-Building by Computer
Molecular Model-building by Computer In which biochemists observe models of giant molecules as they are displayed on a screen by a computer and try to fold them into the shapes that they assume in nature by Cyrus Levinthal any problems of modern biology a large number of problems in biology computer to help gain such information are concerned with the detailed into which biologists have some insight about the structure of large biological M relation between biological but concerning which they cannot yet molecules. function and molecular structure. Some ask suitable questions in terms of molec- For the first half of this century the of the questions currently being asked ular structure. As they see such prob- metabolic and structural relations among will be completely answered only when lems more clearly, however, they in- the small molecules of the living cell one has an understanding of the struc- variably find an increasing need for were the principal concern of bio- ture of all the molecular components of structural information. In our laboratory chemists. The chemical reactions these a biological system and a knowledge of at the Massachusetts Institute of Tech- molecules undergo have been studied how they interact. There are, of course, nology we have recently started using a intensively. Such reactions are specifical- ly catalyzed by the large protein mole- cules called enzymes, many of which have now been purified and also studied. It is only within the past few years, however, that X-ray-diffraction techniques have made it possible to determine the molecular structure of such protein molecules. -

Ontology-Based Classification of Molecules
Ontology-Based Classification of Molecules: a Logic Programming Approach Despoina Magka Department of Computer Science, University of Oxford Wolfson Building, Parks Road, OX1 3QD, UK [email protected] Abstract. We describe a prototype that performs structure-based classification of molecular structures. The software we present implements a sound and com- plete reasoning procedure of a formalism that extends logic programming and builds upon the DLV deductive databases system. We capture a wide range of chemical classes that are not expressible with OWL-based formalisms such as cyclic molecules, saturated molecules and alkanes. In terms of performance, a no- ticeable improvement is observed in comparison with previous approaches. Our evaluation has discovered subsumptions that are missing from the the manually curated ChEBI ontology as well as discrepancies with respect to existing subclass relations. We illustrate thus the potential of an ontology language which is suit- able for the Life Sciences domain and exhibits an encouraging balance between expressive power and practical feasibility. Keywords: Knowledge representation and reasoning, Logic programming and answer set programming, Cheminformatics. 1 Introduction The volume of bioinformatics data produced by research laboratories worldwide is in- creasing at an astonishing rate turning the need to adequately catalogue, represent and index the vast amounts of Life Sciences data sources into a pressing challenge. Semantic technologies have achieved significant progress towards the federation of biochemical information [33, 3, 4] via the definition and use of domain vocabularies with formal semantics, also known as ontologies. OWL [15], a family of logic-based knowledge representation (KR) formalisms standardised by W3C, has played a pivotal role in the advent of Semantic technologies due to its significant ability to reason over ontolo- gies by means of logical inference. -

CORINA Classic Program Manual
CORINA Classic Generation of Three-Dimensional Molecular Models Version 4.4.0 Program Description Molecular Networks GmbH September 2021 www.mn-am.com Molecular Networks GmbH Altamira LLC Neumeyerstraße 28 470 W Broad St, Unit #5007 90411 Nuremberg Columbus, OH 43215 Germany USA mn-am.com This document is copyright © 1998-2021 by Molecular Networks GmbH Computerchemie and Altamira LLC. All rights reserved. Except as permitted under the terms of the Software Licensing Agreement of Molecular Networks GmbH Computerchemie, no part of this publication may be reproduced or distributed in any form or by any means or stored in a database retrieval system without the prior written permission of Molecular Networks GmbH or Altamira LLC. The software described in this document is furnished under a license and may be used and copied only in accordance with the terms of such license. (Document version: 4.4.0-2021-09-30) Content Content 1 Introducing CORINA Classic 1 1.1 Objective of CORINA Classic 1 1.2 CORINA Classic in Brief 1 2 Release Notes 3 2.1 CORINA (Full Version) 3 2.2 CORINA_F (Restricted FlexX Interface Version) 25 3 Getting Started with CORINA Classic 26 4 Using CORINA Classic 29 4.1 Synopsis 29 4.2 Options 29 5 Use Cases of CORINA Classic 60 6 Supported File Formats and Interfaces 64 6.1 V2000 Structure Data File (SD) and Reaction Data File (RD) 64 6.2 V3000 Structure Data File (SD) and Reaction Data File (RD) 67 6.3 SMILES Linear Notation 67 6.4 InChI file format 69 6.5 SYBYL File Formats 70 6.6 Brookhaven Protein Data Bank Format (PDB) -

(CDK): an Open-Source Java Library for Chemo- and Bioinformatics
J. Chem. Inf. Comput. Sci. 2003, 43, 493-500 493 The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics Christoph Steinbeck,*,† Yongquan Han,† Stefan Kuhn,† Oliver Horlacher,‡ Edgar Luttmann,§ and Egon Willighagen# Max-Planck-Institute of Chemical Ecology, Jena, Germany, TheraSTrat AG, Allschwil, Switzerland, Institute of Organic Chemistry, University of Paderborn, Germany, and Nijmegen, The Netherlands Received August 17, 2002 The Chemistry Development Kit (CDK) is a freely available open-source Java library for Structural Chemo- and Bioinformatics. Its architecture and capabilities as well as the development as an open-source project by a team of international collaborators from academic and industrial institutions is described. The CDK provides methods for many common tasks in molecular informatics, including 2D and 3D rendering of chemical structures, I/O routines, SMILES parsing and generation, ring searches, isomorphism checking, structure diagram generation, etc. Application scenarios as well as access information for interested users and potential contributors are given. 1. INTRODUCTION of software development, most widely recognized through the great success of the free Unix-like operating system Whoever pursues the endeavor of creating a larger GNU/Linux, a collaborative work of many individuals and software package in chemoinformatics or computational organizations, including the Free Software Foundation lead chemistry from scratch will soon be confronted with the by Richard Stallman and the Finish computer science student Syssiphus task of implementing the standard repertoire of Linus Torvalds who started the project. According to several chemoinformatical algorithms and components invented essays on this subject, open-source software, for which, by during the last 20 or 30 years. -

Chemical File Format Conversion Tools : a N Overview
International Journal of Engineering Research & Technology (IJERT) ISSN: 2278-0181 Vol. 3 Issue 2, February - 2014 Chemical File Format Conversion Tools : A n Overview Kavitha C. R Dr. T Mahalekshmi Research Scholar, Bharathiyar University Principal Dept of Computer Applications Sree Narayana Institute of Technology SNGIST Kollam, India Cochin, India Abstract— There are a lot of chemical data stored in large different chemical file formats. Three types of file format databases, repositories and other resources. These data are used conversion tools are discussed in section III. And the by different researchers in different applications in various areas conclusion is given in section IV followed by the references. of chemistry. Since these data are stored in several standard chemical file formats, there is a need for the inter-conversion of II. CHEMICAL FILE FORMATS chemical structures between different formats because all the formats are not supported by various software and tools used by A chemical is a collection of atoms bonded together the researchers. Therefore it becomes essential to convert one file in space. The structure of a chemical makes it unique and format to another. This paper reviews some of the chemical file gives it its physical and biological characteristics. This formats and also presents a few inter-conversion tools such as structure is represented in a variety of chemical file formats. Open Babel [1], Mol converter [2] and CncTranslate [3]. These formats are used to represent chemical structure records and its associated data fields. Some of the file Keywords— File format Conversion, Open Babel, mol formats are CML (Chemical Markup Language), SDF converter, CncTranslate, inter- conversion tools.