CORINA Classic Program Manual
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -
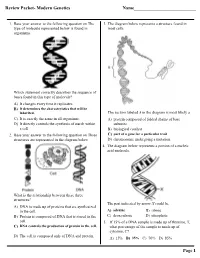
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Application of X-Ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-19-2008 Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis Zakhia Moore University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Moore, Zakhia, "Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis" (2008). University of New Orleans Theses and Dissertations. 888. https://scholarworks.uno.edu/td/888 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Chemistry by Zakhia Moore B.S., Xavier University, 2000 M.S., University of New Orleans, 2005 December, 2008 ACKNOWLEDGEMENTS Giving all glory to God for His many blessings, whom without I could not have completed this long journey. -

Decalin Dehydrogenation for In-Situ Hydrogen Production To
DECALIN DEHYDROGENATION FOR IN-SITU HYDROGEN PRODUCTION TO INCREASE CATALYTIC CRACKING RATE OF N-DODECANE Thesis Submitted to The School of Engineering of the UNIVERSITY OF DAYTON In Partial Fulfillment of the Requirements for The Degree of Master of Science in Chemical Engineering By Christopher Bruening Dayton, Ohio May, 2018 DECALIN DEHYDROGENATION FOR IN-SITU HYDROGEN PRODUCTION TO INCREASE CATALYTIC CRACKING RATE OF N-DODECANE Name: Bruening, Christopher Robbins APPROVED BY: Matthew J. DeWitt, Ph.D. Donald K. Phelps, Ph.D. Advisory Committee Chairman Committee Member Distinguished Research Engineer Senior Research Chemist University of Dayton Research Institute Air Force Research Laboratory Michael Elsass, Ph.D. Kevin Myers, D.Sc., P.E. Committee Member Committee Member Lecturer Professor Department of Chemical and Materials Department of Chemical and Materials Engineering Engineering Robert J. Wilkens, Ph.D., P.E. Eddy M. Rojas, Ph.D., M.A., P.E. Associate Dean for Research and Innovation Dean, School of Engineering Professor School of Engineering School of Engineering ii ABSTRACT DECALIN DEHYDROGENATION FOR IN-SITU HYDROGEN PRODUCTION TO INCREASE CATALYTIC CRACKING RATE OF N-DODECANE Name: Bruening, Christopher Robbins University of Dayton Advisor: Dr. Matthew J. DeWitt Catalytic cracking of paraffinic hydrocarbons is a widely utilized industrial process, but catalyst deactivation over time requires regeneration or replacement of the catalyst bed. A gaseous hydrogen co-feed can be used to promote hydrocracking and decrease deactivation of the catalyst due to coke formation or active site poisoning. One potential alternative approach to extend the lifetime of a cracking catalyst is to generate molecular hydrogen in-situ via catalytic dehydrogenation of a cycloparaffin. -

Dibasic Acids for Nylon Manufacture
- e Report No. 75 DIBASIC ACIDS FOR NYLON MANUFACTURE by YEN-CHEN YEN October 1971 A private report by the PROCESS ECONOMICS PROGRAM STANFORD RESEARCH INSTITUTE MENLO PARK, CALIFORNIA CONTENTS INTRODUCTION, ....................... 1 SUMMARY .......................... 3 General Aspects ...................... 3 Technical Aspects ..................... 7 INDUSTRY STATUS ...................... 15 Applications and Consumption of Sebacic Acid ........ 15 Applications and Consumption of Azelaic Acid ........ 16 Applications of Dodecanedioic and Suberic Acids ...... 16 Applications of Cyclododecatriene and Cyclooctadiene .... 17 Producers ......................... 17 Prices ........................... 18 DIBASIC ACIDS FOR MANUFACTURE OF POLYAMIDES ........ 21 CYCLOOLIGOMERIZATIONOF BUTADIENE ............. 29 Chemistry ......................... 29 Ziegler Catalyst ..................... 30 Nickel Catalyst ..................... 33 Other Catalysts ..................... 34 Co-Cyclooligomerization ................. 34 Mechanism ........................ 35 By-products and Impurities ................ 37 Review of Processes .................... 38 A Process for Manufacture of Cyclododecatriene ....... 54 Process Description ................... 54 Process Discussion .................... 60 Cost Estimates ...................... 60 A Process for Manufacture of Cyclooctadiene ........ 65 Process Description ................... 65 Process Discussion .................... 70 Cost Estimates ...................... 70 A Process for Manufacture of Cyclodecadiene -

Organic Contaminant Analytical Methods of the National Status and Trends Program: 2000-2006
Organic Contaminant Analytical Methods of the National Status and Trends Program: 2000-2006 NOAA Technical Memorandum NOS NCCOS 30 Mention of trade names or commercial products does not constitute endorsement or recommendation for their use by the United States government. Cover photograph of shrimping fleet, Palacios, TX Citation for this Report Kimbrough, K. L., G. G. Lauenstein and W. E. Johnson (Editors). 2006. Organic Contaminant Analytical Methods of the National Status and Trends Program: Update 2000-2006. NOAA Technical Memorandum NOS NCCOS 30. 137 pp. Organic Contaminant Analytical Methods of the National Status and Trends Program: Update 2000-2006 K. L. Kimbrough, G. G. Lauenstein and W. E. Johnson (Editors) Center for Coastal Monitoring and Assessment NOAA/NOS/NCCOS 1305 East-West Highway Silver Spring, Maryland 20910 NOAA Technical Memorandum NOS NCCOS 30 June 2007 United States Department of National Oceanic and National Ocean Service Commerce Atmospheric Administration Carlos M. Gutierrez Conrad C. Lautenbacher, Jr. John H. Dunnigan Secretary Administrator Assistant Administrator TABLE OF CONTENTS TABLE OF CONTENTS ..................................................................................................I LIST OF TABLES ..........................................................................................................III LIST OF FIGURES ........................................................................................................IV CHAPTER 1. INTRODUCTION................................................................................... -

Research on Crystal Growth and Characterization at the National Bureau of Standards January to June 1964
NATL INST. OF STAND & TECH R.I.C AlllDS bnSflb *^,; National Bureau of Standards Library^ 1*H^W. Bldg Reference book not to be '^sn^ t-i/or, from the library. ^ecknlccil v2ote 251 RESEARCH ON CRYSTAL GROWTH AND CHARACTERIZATION AT THE NATIONAL BUREAU OF STANDARDS JANUARY TO JUNE 1964 U. S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS tiona! Bureau of Standards NOV 1 4 1968 151G71 THE NATIONAL BUREAU OF STANDARDS The National Bureau of Standards is a principal focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. Its responsibilities include development and maintenance of the national stand- ards of measurement, and the provisions of means for making measurements consistent with those standards; determination of physical constants and properties of materials; development of methods for testing materials, mechanisms, and structures, and making such tests as may be necessary, particu- larly for government agencies; cooperation in the establishment of standard practices for incorpora- tion in codes and specifications; advisory service to government agencies on scientific and technical problems; invention and development of devices to serve special needs of the Government; assistance to industry, business, and consumers in the development and acceptance of commercial standards and simplified trade practice recommendations; administration of programs in cooperation with United States business groups and standards organizations for the development of international standards of practice; and maintenance of a clearinghouse for the collection and dissemination of scientific, tech- nical, and engineering information. The scope of the Bureau's activities is suggested in the following listing of its four Institutes and their organizational units. -

IAPS Act 15-17 Section Preview of the Teachers Edition.Pdf
Section Preview of the Teacher’s Edition for The Chemistry of Materials, Issues and Physical Science, 2nd Edition Activities 15-17 Suggested student responses and answer keys have been blocked out so that web-savvy students do not find this page and have access to answers. To experience a complete activity please request a sample found in the footer at lab-aids.com Families of Elements 15 4 s 0 2 n - o t si o es 50 te s -minu N O I T I N A ACTIVITY OVERVIEW V E S T I G Students compare physical and chemical properties of 13 elements and sort them into groups based on common properties. They then compare their classifications with groups—or families—of elements as defined by scientists and displayed in the Periodic Table of the Elements. KEY CONCEPTS AND PROCESS SKILLS (with correlation to NSE 5–8 Content Standards) 1. Elements have characteristic physical and chemical properties. Substances often are placed in categories or groups if they react in similar ways; metals are an example of such a group. (PhysSci: 1) 2. Scientists classify elements into families, based on their properties. (PhysSci: 1) 3. Students develop descriptions based on evidence. (Inquiry: 1) KEY VOCABULARY atom atomic mass element family (of elements) metal Periodic Table of the Elements B-39 Activity 15 • Families of Elements MATERIALS AND ADVANCE PREPARATION For the teacher aluminum, iron, copper, and carbon samples from Activity 14, “Physical and Chemical Properties of Materials” 1 Transparency 15.1, “Information on Element Cards” 8 sets of 4 Element Family Cards -

BOSS, Version 4.6 Biochemical and Organic Simulation System User’S Manual for UNIX, Linux, and Windows
BOSS, Version 4.6 Biochemical and Organic Simulation System User’s Manual for UNIX, Linux, and Windows William L. Jorgensen Department of Chemistry, Yale University P. O. Box 208107 New Haven, Connecticut 06520-8107 Phone: (203) 432-6278 Fax: (203) 432-6299 Internet: [email protected] January 2004 Contents Page Introduction 4 1 Can’t Wait to Start – Use the x Scripts 6 2 Statistical Mechanics Simulation – Theory 6 3 Energy and Free Energy Evaluation 7 4 New Features 9 5 Operating Systems 14 6 Installation 14 7 Files 14 8 Command or bat File Input 16 9 Parameter File Input 18 10 Z-matrix File Input 26 10.1 Atom Input 29 10.2 Geometry Variations 29 10.3 Bond Length Variations 29 10.4 Additional Bonds 30 10.5 Harmonic Restraints 30 10.6 Bond Angle Variations 30 10.7 Additional Bond Angles 31 10.8 Dihedral Angle Variations 31 10.9 Additional Dihedral Angles 33 10.10 Domain Definitions 34 10.11 Conformational Searching 34 10.12 Sample Z-matrix 34 10.13 Z-matrix Input for Custom Solvents 35 11 PDB Input 37 12 Coordinate Input in Mind Format and Reaction Path Following 38 13 Pure Liquid Simulations 39 14 Cluster Simulations 39 2 15 Solventless and Molecular Mechanics Calculations 40 15.1 Continuum Simulations 40 15.2 Energy Minimizations 41 15.3 Dihedral Angle Driving 42 15.4 Potential Surface Scanning 42 15.5 Normal Coordinate Analysis 43 15.6 Conformational Searching 45 16 Available Solvent Boxes 48 17 Ewald Summation for Long-Range Electrostatics 50 18 Test Jobs 51 19 Output 56 19.1 Some Variable Definitions 58 20 Contents of the Distribution Files 59 21 Appendix – No. -

United States Patent Office Patented
2,826,604 United States Patent Office Patented. Mar. 11, 1958 2 (130-140 C.), the decalyl acetate being recoverable from the reaction mixture by distillation of the neutralized re 2,826,604 action liquid. The recovered product was a straw-colored PREPARATION OF DECAHYDRONAPHTHYL fragrant oil, which was established to be 1-decallyl acetate. ACETATE 5 The position of the acetoxy group was determined by William E. Erner, Wilmington, Del, assignor to Houdry hydrolysis of the ester to the alcohol and subsequent Process Corporation, Wilmington, Del, a corporation conversion to decalone-1 by chromic acid-acetic acid oxi of Delaware dation. - The aceto-oxidation of Decalin can be carried out No Drawing. Application June 2, 1955 10 in the presence of oxidation catalysts of the type em Serial No. 512,853 bodied in the metal salts of the lower fatty acids. Cobalt 2 Claims. (C. 260-488) acetate proved outstanding among the metallic-soap oxi dation catalysts studied. The use of benzoyl peroxide at the start of the process has been found advantageous The present invention relates to the preparation of 1 5 in decreasing the induction period and in giving uni decahydronaphthyl alcohols and the corresponding acetic formly high yields. acid ester and ketone derivatives of Said alcohols. The ratios of Decalin - to acetic anhydride in the oper More particularly the invention is concerned with the ation of the process are not critical, but practical con production of saturated polynuclear carbocyclic com siderations favor the use of at least one-half mol of pounds of the type 20 acetic anhydride per mol of Decalin. -

Molecular Model-Building by Computer
Molecular Model-building by Computer In which biochemists observe models of giant molecules as they are displayed on a screen by a computer and try to fold them into the shapes that they assume in nature by Cyrus Levinthal any problems of modern biology a large number of problems in biology computer to help gain such information are concerned with the detailed into which biologists have some insight about the structure of large biological M relation between biological but concerning which they cannot yet molecules. function and molecular structure. Some ask suitable questions in terms of molec- For the first half of this century the of the questions currently being asked ular structure. As they see such prob- metabolic and structural relations among will be completely answered only when lems more clearly, however, they in- the small molecules of the living cell one has an understanding of the struc- variably find an increasing need for were the principal concern of bio- ture of all the molecular components of structural information. In our laboratory chemists. The chemical reactions these a biological system and a knowledge of at the Massachusetts Institute of Tech- molecules undergo have been studied how they interact. There are, of course, nology we have recently started using a intensively. Such reactions are specifical- ly catalyzed by the large protein mole- cules called enzymes, many of which have now been purified and also studied. It is only within the past few years, however, that X-ray-diffraction techniques have made it possible to determine the molecular structure of such protein molecules. -

Macromodel Quick Start Guide
MacroModel Quick Start Guide MacroModel 9.9 Quick Start Guide Schrödinger Press MacroModel Quick Start Guide Copyright © 2012 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication, Schrödinger assumes no responsibility for errors or omissions, or for damages resulting from the use of the information contained herein. BioLuminate, Canvas, CombiGlide, ConfGen, Epik, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Phase, Prime, PrimeX, QikProp, QikFit, QikSim, QSite, SiteMap, Strike, and WaterMap are trademarks of Schrödinger, LLC. Schrödinger and MacroModel are registered trademarks of Schrödinger, LLC. MCPRO is a trademark of William L. Jorgensen. DESMOND is a trademark of D. E. Shaw Research, LLC. Desmond is used with the permission of D. E. Shaw Research. All rights reserved. This publication may contain the trademarks of other companies. Schrödinger software includes software and libraries provided by third parties. For details of the copyrights, and terms and conditions associated with such included third party software, see the Legal Notices, or use your browser to open $SCHRODINGER/docs/html/third_party_legal.html (Linux OS) or %SCHRODINGER%\docs\html\third_party_legal.html (Windows OS). This publication may refer to other third party software not included in or with Schrödinger software ("such other third party software"), and provide links to third party Web sites ("linked sites"). References to such other third party software or linked sites do not constitute an endorsement by Schrödinger, LLC or its affiliates. Use of such other third party software and linked sites may be subject to third party license agreements and fees. Schrödinger, LLC and its affiliates have no responsibility or liability, directly or indirectly, for such other third party software and linked sites, or for damage resulting from the use thereof.