Herman Skolnik Award Symposium 2016
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Open Babel Documentation Release 2.3.1
Open Babel Documentation Release 2.3.1 Geoffrey R Hutchison Chris Morley Craig James Chris Swain Hans De Winter Tim Vandermeersch Noel M O’Boyle (Ed.) December 05, 2011 Contents 1 Introduction 3 1.1 Goals of the Open Babel project ..................................... 3 1.2 Frequently Asked Questions ....................................... 4 1.3 Thanks .................................................. 7 2 Install Open Babel 9 2.1 Install a binary package ......................................... 9 2.2 Compiling Open Babel .......................................... 9 3 obabel and babel - Convert, Filter and Manipulate Chemical Data 17 3.1 Synopsis ................................................. 17 3.2 Options .................................................. 17 3.3 Examples ................................................. 19 3.4 Differences between babel and obabel .................................. 21 3.5 Format Options .............................................. 22 3.6 Append property values to the title .................................... 22 3.7 Filtering molecules from a multimolecule file .............................. 22 3.8 Substructure and similarity searching .................................. 25 3.9 Sorting molecules ............................................ 25 3.10 Remove duplicate molecules ....................................... 25 3.11 Aliases for chemical groups ....................................... 26 4 The Open Babel GUI 29 4.1 Basic operation .............................................. 29 4.2 Options ................................................. -

JRC QSAR Model Database
JRC QSAR Model Database EURL ECVAM DataBase service on ALternative Methods to animal experimentation To promote the development and uptake of alternative and advanced methods in toxicology and biomedical sciences SDF - STRUCTURE DATA FORMAT: How to create from SMILES The European Commission’s science and knowledge service Joint Research Centre Directorate F Health, Consumers & Reference Materials Chemicals Safety & Alternative Methods Unit The European Commission’s science and knowledge service Joint Research Centre EUR 28708 EN This publication is a Tutorial by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide user support. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Email: [email protected] JRC Science Hub https://ec.europa.eu/jrc JRC107492 EUR 28708 EN PDF ISBN 978-92-79-71294-4 ISSN 1831-9424 doi:10.2760/952280 Print ISBN 978-92-79-71295-1 ISSN 1018-5593 doi:10.2760/668595 Luxembourg: Publications Office of the European Union, 2017 Ispra: European Commission, 2017 © European Union, 2017 The reuse of the document is authorised, provided the source is acknowledged and the original meaning or message of the texts are not distorted. The European Commission shall not be held liable for any consequences stemming from the reuse. How to cite this document: Triebe -
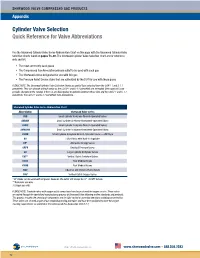
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Colorimetric Sugar Sensing Using Boronic Acid-Substituted Azobenzenes
Materials 2014, 7, 1201-1220; doi:10.3390/ma7021201 OPEN ACCESS materials ISSN 1996-1944 www.mdpi.com/journal/materials Review Colorimetric Sugar Sensing Using Boronic Acid-Substituted Azobenzenes Yuya Egawa *, Ryotaro Miki and Toshinobu Seki Faculty of Pharmaceutical Sciences, Josai University, Keyakidai, Sakado, Saitama 350-0295, Japan; E-Mails: [email protected] (R.M.); [email protected] (T.S.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-49-271-7686; Fax: +81-49-271-7714. Received: 30 November 2013; in revised form: 13 January 2014 / Accepted: 28 January 2014 / Published: 14 February 2014 Abstract: In association with increasing diabetes prevalence, it is desirable to develop new glucose sensing systems with low cost, ease of use, high stability and good portability. Boronic acid is one of the potential candidates for a future alternative to enzyme-based glucose sensors. Boronic acid derivatives have been widely used for the sugar recognition motif, because boronic acids bind adjacent diols to form cyclic boronate esters. In order to develop colorimetric sugar sensors, boronic acid-conjugated azobenzenes have been synthesized. There are several types of boronic acid azobenzenes, and their characteristics tend to rely on the substitute position of the boronic acid moiety. For example, o-substitution of boronic acid to the azo group gives the advantage of a significant color change upon sugar addition. Nitrogen-15 Nuclear Magnetic Resonance (NMR) studies clearly show a signaling mechanism based on the formation and cleavage of the B–N dative bond between boronic acid and azo moieties in the dye. -

Study of Photo-Induced Dichroism in Sudan III Doped in Poly(Methyl Methacrylate) Thin Films B
Vol. 127 (2015) ACTA PHYSICA POLONICA A No. 3 Study of Photo-Induced Dichroism in Sudan III Doped in Poly(Methyl Methacrylate) Thin Films B. Abbasa;* and Y.T. Salmanb aAtomic Energy Commission, P.O. Box 6091, Damascus, Syria bPhysics Department, Faculty of Science, Damascus University, Damascus, Syria (Received December 4, 2013; in nal form December 10, 2014) Linear dichroism, dichroic ratio, contrast ratio and order parameters of Sudan III/PMMA guesthost thin lms have been investigated with visible polarized laser light. Dichroism increased in an exponential fashion with increase of the pump intensity. Light-induced dichroism and polar order of the dye molecules within the polymer network were reversible. These parameters showed fast increase in their values in an exponential fashion as the pumping process takes place. Also, they decay rapidly in an exponential fashion when the pump light is cut o. Photoisomerisation and polar orientation are dependent on both the molecular structure of the dye and polymer. Two forms of Sudan III molecules (trans and cis) are responsible for the interaction with laser light and forming an anisotropic structure inside the PMMA/Sudan III lms. However, there is another set of forms of the dye (keto and enol) has to be considered, which may contribute to dichroism. DOI: 10.12693/APhysPolA.127.780 PACS: 78.20.Ek, 33.55.+b, 78.66.Qn 1. Introduction Sudan III in solvents and doped in polymeric lms using the Z-scan technique. Multiple diraction rings were ob- Photoinduced optical anisotropy in polymer/organic served when the dye doped in both the liquid and solid dyes systems is the subject of intensive care and inves- media was exposed to a diode-pumped Nd:YAG laser tigations [1]. -

Evolution of Refrigerants Mark O
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jced Review (R)Evolution of Refrigerants Mark O. McLinden* and Marcia L. Huber Cite This: J. Chem. Eng. Data 2020, 65, 4176−4193 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: As we enter the “fourth generation” of refrigerants, we consider the evolution of refrigerant molecules, the ever- changing constraints and regulations that have driven the need to consider new molecules, and the advancements in the tools and property models used to identify new molecules and design equipment using them. These separate aspects are intimately intertwined and have been in more-or-less continuous development since the earliest days of mechanical refrigeration, even if sometimes out-of-sight of the mainstream refrigeration industry. We highlight three separate, comprehensive searches for new refrigerantsin the 1920s, the 1980s, and the 2010sthat sometimes identified new molecules, but more often, validated alternatives already under consideration. A recurrent theme is that there is little that is truly new. Most of the “new” refrigerants, from R-12 in the 1930s to R- 1234yf in the early 2000s, were reported in the chemical literature decades before they were considered as refrigerants. The search for new refrigerants continued through the 1990s even as the hydrofluorocarbons (HFCs) were becoming the dominant refrigerants in commercial use. This included a return to several long-known natural refrigerants. Finally, we review the evolution of the NIST REFPROP database for the calculation of refrigerant properties. -

MACSIMUS Manual
1 MACSIMUS manual benzocaine (ethylaminobenzoate) parameter_set = charmm21 HA | HA-CT-HA | HA-CT-HA | OSn.2 | Cp.7=On.5 | C6R--C6R-HA Most often used links: | | HA-C6R C6R-HA 2.2 Blend synopsis and options | | HA-C6R--C6R-NPn.5^-Hp.25 9.2 Cook synopsis and options | 9.2.5 Cook input data Hp.25 MACromolecule SIMUlation Software © Jiˇr´ıKolafa 1993{2020 MACSIMUS may be distributed under the terms of the GNU General Public Licence Credits: ray: Mark VandeWettering \reasonably intelligent raytracer" CHARMM force field (files: charmm*.par, charmm*/*.rsd) GROMOS force field (files: gromos*.par, gromos*/*.rsd) amoeba implementation by Z. Wagner moil support by J. Schofield several bug fixes by T. Trnka bug discovered by N. Parfenov Contents I Program `blend' version 2.4b 14 1 Introduction 16 1.1 Force fields...................................... 16 1.2 `blend' overview.................................... 16 1.3 Versions........................................ 17 2 Running blend 18 2.1 Environment...................................... 18 2.2 Synopsis........................................ 19 2.2.1 Global options................................. 19 2.2.2 par-options and parameter files....................... 20 2.2.3 mol-options and molecular files....................... 22 2.2.4 Extra-options ................................. 29 2.3 File extensions.................................... 34 2.4 Run-time control................................... 37 2.4.1 get data format for input........................... 37 2.4.2 Scrolling.................................... 38 2.4.3 Error handling................................ 39 2.4.4 Interrupts................................... 40 2.5 Showing molecules graphically............................ 40 2.5.1 X11 Graphics................................. 40 2.5.2 Playback output............................... 44 2.6 Energy minimization................................. 44 2.7 Missing coordinates.................................. 45 3 Force field and the parameter file 46 3.1 Structure of the parameter file........................... -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

Studying Azobenzene-Modified DNA for Programmable Nanoparticle Assembly and Nucleic Acid Detection
Studying Azobenzene-Modified DNA for Programmable Nanoparticle Assembly and Nucleic Acid Detection Yunqi Yan A dissertation submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee David Ginger, Chair Daniel Chiu Lutz Maibaum Program Authorized to Offer Degree: Chemistry 1 ©Copyright 2015 Yunqi Yan 2 University of Washington Abstract Studying Azobenzene-Modified DNA for Programmable Nanoparticle Assembly and Nucleic Acid Detection Yunqi Yan Chair of the Supervisory Committee: Professor David Ginger Department of Chemistry The azobenzene-modified DNA is an oligonucleotide molecule with azobenzene moieties covalently linked to the DNA backbone via the d-threoninol bond. The reversible photoswitching capability of azobenzene allows optical irradiation (light) to control the hybridization of azobenzene-modified DNA. Meanwhile, gold nanoparticles functionalized with natural oligonucleotides show increased sensitivity in nucleic acid detection due to the DNA-directed biorecognition and nanoparticles’ plasmonic effects. In the dissertation, I present my studies of surface functionalization of gold nanoparticles with azobenzene-modified oligonucleotides and the relevant photochemical characterization of the new system. In chapter 2, I synthesize the photoswichable gold nanoparticle assemblies cross-linked with azobenzene-modified DNA. Beyond the classic DNA-directed assembly and sensing behaviors associated with DNA-modified nanoparticles, these particles exhibit reversible photoswitching of their assembly behavior. Exposure to UV light induces the dissociation of nanoparticle aggregates due to trans-to-cis isomerization of the azobenzene which destabilizes the DNA duplex. The assembly of nanoparticles is reversible upon exposure to blue light due to the reverse cis-to-trans isomerization of azobenzene-modified DNA. -

THOMAS MIDGLEY, JR., and the INVENTION of CHLOROFLUOROCARBON REFRIGERANTS: IT AIN’T NECESSARILY SO Carmen J
66 Bull. Hist. Chem., VOLUME 31, Number 2 (2006) THOMAS MIDGLEY, JR., AND THE INVENTION OF CHLOROFLUOROCARBON REFRIGERANTS: IT AIN’T NECESSARILY SO Carmen J. Giunta, Le Moyne College The 75th anniversary of the first public Critical readers are well aware description of chlorofluorocarbon (CFC) of the importance of evaluating refrigerants was observed in 2005. A sources of information. For ex- symposium at a national meeting of ample, an article in Nature at the the American Chemical Society (ACS) end of 2005 tested the accuracy on CFCs from invention to phase-out of two encyclopedias’ entries on a (1) and an article on their invention sample of topics about science and and inventor in the Chemical Educa- history of science (3). A thought- tor (2) marked the occasion. The story ful commentary published soon of CFCs—their obscure early days as afterwards raised questions on just laboratory curiosities, their commercial what should count as an error in debut as refrigerants, their expansion into assessing such articles: omissions? other applications, and the much later disagreements among generally re- discovery of their deleterious effects on liable sources (4)? Readers of his- stratospheric ozone—is a fascinating torical narratives who are neither one of science and society well worth practicing historians nor scholarly telling. That is not the purpose of this amateurs (the category to which I article, though. aspire) may find an account of a Thomas Midgley, Jr. historical research process attrac- This paper is about contradictory Courtesy Richard P. Scharchburg tive. As a starting point, consider sources, foggy memories, the propaga- Archives, Kettering University the following thumbnail summary tion of error, and other obstacles to writ- of the invention. -

Overview of Fluorocarbons
Three Bond Technical News Issued Dec. 20, 1989 An Overview of Fluorocarbons Introduction Currently, chlorofluorocarbons are garnering a lot of Three Bond Co,. Ltd. has been working night and day in attention around the world. Chlorofluorocarbons are used in search of alternatives to chlorofluorocarbons. In the future, many of our products and when we look at the future of products that do not contain any chlorofluorocarbons will chlorofluorocarbons in the world, and in Japan, we can replace products that now contain chlorofluorocarbons. To foresee that their use will be strictly limited or eliminated all ensure these products reach our customers as smoothly as together. possible, it is important that they have a good understanding of the issues surrounding chlorofluorocarbons. Whatever the case, Three Bond Co., Ltd. is obligated to provide our customers with products that have the same functionality as chlorofluorocarbons. Contents Introduction ................................................................................................................................................................1 1. What is a fluorocarbon? .......................................................................................................................................2 2. Properties of fluorocarbons ..................................................................................................................................2 3. Types of fluorocarbons.........................................................................................................................................2 -

Ontology-Based Classification of Molecules
Ontology-Based Classification of Molecules: a Logic Programming Approach Despoina Magka Department of Computer Science, University of Oxford Wolfson Building, Parks Road, OX1 3QD, UK [email protected] Abstract. We describe a prototype that performs structure-based classification of molecular structures. The software we present implements a sound and com- plete reasoning procedure of a formalism that extends logic programming and builds upon the DLV deductive databases system. We capture a wide range of chemical classes that are not expressible with OWL-based formalisms such as cyclic molecules, saturated molecules and alkanes. In terms of performance, a no- ticeable improvement is observed in comparison with previous approaches. Our evaluation has discovered subsumptions that are missing from the the manually curated ChEBI ontology as well as discrepancies with respect to existing subclass relations. We illustrate thus the potential of an ontology language which is suit- able for the Life Sciences domain and exhibits an encouraging balance between expressive power and practical feasibility. Keywords: Knowledge representation and reasoning, Logic programming and answer set programming, Cheminformatics. 1 Introduction The volume of bioinformatics data produced by research laboratories worldwide is in- creasing at an astonishing rate turning the need to adequately catalogue, represent and index the vast amounts of Life Sciences data sources into a pressing challenge. Semantic technologies have achieved significant progress towards the federation of biochemical information [33, 3, 4] via the definition and use of domain vocabularies with formal semantics, also known as ontologies. OWL [15], a family of logic-based knowledge representation (KR) formalisms standardised by W3C, has played a pivotal role in the advent of Semantic technologies due to its significant ability to reason over ontolo- gies by means of logical inference.