"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UNITED STATES PATENT Office SVEND S
Patented May 24, 1932 1,859,998 UNITED STATES PATENT oFFICE SVEND S. SVENDSEN, OF CHICAGO, ILLINOIS, ASSIGNOR TO CLAY REDUCTION comi PANY, OF CHICAGO, ILLINOIS, A CORPORATION OF ILLINOIs . HYDRATED SILICA No Drawing. Application filed December 23, 1927. Serial No. 242,291. This invention relates to the production of silicofluoride is also volatilized and collected hydrated silica from a silicious material such in aqueous animonia. The volatilization oc as silica and silicates generally. - - - - curs about 300 C. The following reactions According to the invention the silicious take place in the aqueous ammonia at tem 5 material is treated with ammonium fluoride peratures below 34 C.: 5 5 or bifluoride and silicon ammonia fluorine compounds are produced. These compounds 2SiF(NH) +2H.O= are volatilized and converted into hydrated (NH4)2SiFs--SiO, (hydrated) + silica by the action of water and ammonia. 2NH.F. (IV) O Metallic silicates or substances containing (NH)SiF+4NH,+2HO = them can be subjected to this treatment di SiO, (hydrated) + 6NH.F. (V) rectly. In treating silica in the form of quartz it is found to be necessary to subject It is thus apparent that O heating the it to a preliminary treatment in order to fa silicious material with the ammonium-flu cilitate the action of the ammonium fluoride oride, ammonia-silicon-fluorine compounds a or bifluoride thereon. A suitable preliminary are formed, and by employing suitable tem treatment is to heat the quartz to a bright red peratures are volatilized from the reaction heat and suddenly cool it by immersion in mixture. These compounds may be silicon water. -

Basic Aspects of Fluorine in Chemistry and Biology
Introduction: Basic Aspects of Fluorine in Chemistry and Biology COPYRIGHTED MATERIAL 1 Unique Properties of Fluorine and Their Relevance to Medicinal Chemistry and Chemical Biology Takashi Yamazaki , Takeo Taguchi , and Iwao Ojima 1.1 Fluorine - Substituent Effects on the Chemical, Physical and Pharmacological Properties of Biologically Active Compounds The natural abundance of fl uorine as fl uorite, fl uoroapatite, and cryolite is considered to be at the same level as that of nitrogen on the basis of the Clarke number of 0.03. However, only 12 organic compounds possessing this special atom have been found in nature to date (see Figure 1.1) [1] . Moreover, this number goes down to just fi ve different types of com- pounds when taking into account that eight ω - fl uorinated fatty acids are from the same plant [1] . [Note: Although it was claimed that naturally occurring fl uoroacetone was trapped as its 2,4 - dinitrohydrazone, it is very likely that this compound was fl uoroacetal- dehyde derived from fl uoroacetic acid [1] . Thus, fl uoroacetone is not included here.] In spite of such scarcity, enormous numbers of synthetic fl uorine - containing com- pounds have been widely used in a variety of fi elds because the incorporation of fl uorine atom(s) or fl uorinated group(s) often furnishes molecules with quite unique properties that cannot be attained using any other element. Two of the most notable examples in the fi eld of medicinal chemistry are 9α - fl uorohydrocortisone (an anti - infl ammatory drug) [2] and 5 - fl uorouracil (an anticancer drug) [3], discovered and developed in 1950s, in which the introduction of just a single fl uorine atom to the corresponding natural products brought about remarkable pharmacological properties. -

Downloads/DL Praevention/Fachwissen/Gefahrstoffe/TOXIKOLOGI SCHE BEWERTUNGEN/Bewertungen/Toxbew072-L.Pdf
Distribution Agreement In presenting this thesis or dissertation as a partial fulfillment of the requirements for an advanced degree from Emory University, I hereby grant to Emory University and its agents the non-exclusive license to archive, make accessible, and display my thesis or dissertation in whole or in part in all forms of media, now or hereafter known, including display on the world wide web. I understand that I may select some access restrictions as part of the online submission of this thesis or dissertation. I retain all ownership rights to the copyright of the thesis or dissertation. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation. Signature: _____________________________ ______________ Jedidiah Samuel Snyder Date Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Master of Public Health Global Environmental Health _________________________________________ P. Barry Ryan, Ph.D. Committee Chair _________________________________________ Eugene Demchuk, Ph.D. Committee Member _________________________________________ Paige Tolbert, Ph.D. Committee Member Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Bachelor of Science in Engineering, B.S.E. The University of Iowa 2010 Thesis Committee Chair: P. Barry Ryan, Ph.D. An abstract of A thesis submitted to the Faculty of the Rollins School of Public Health of Emory University in partial fulfillment of the requirements for the degree of Master of Public Health in Global Environmental Health 2015 Abstract Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. -

Concentrated Stable Fluorchemical Aqueous Emulsions
Europaisches Patentamt © J European Patent Office © Publication number: 0 282 949 Office europeen des brevets A2 © EUROPEAN PATENT APPLICATION (y Application number: 88104014.1 © mt. ci.«: A61K 9/00 , A61 K 9/50 , A61 K 47/00 © Date of filing: 14.03.88 © Priority: 20.03.87 US 28521 © Applicant: AIR PRODUCTS AND CHEMICALS, INC. © Date of publication of application: Route no. 222 21.09.88 Bulletin 88/38 Trexlertown Pennsylvania 18087(US) © Designated Contracting States: © Inventor: Schweighardt, Frank Kenneth BE CH DE ES FR GB IT Li NL SE 509 Bastian Lane Rd No. 3 Allentown, PA 18104(US) Inventor: Kayhart, Charles Randall Baidy Hill Road P.O.B. 195 Alburtis, PA 18011 (US) © Representative: Dipi.-lng. Schwabe, Dr. Dr. Sandmair, Dr. Marx Stuntzstrasse 16 D-8000 MUnchen 80(DE) © Concentrated stable fluorchemical aqueous emulsions. © A stable concentrated aqueous emulsion of perfluorochemical, a phospholipid and a triglyceride of fatty acids has been demonstrated which has enhanced stability, diminished particle size and heightened tolerance by biological systems. The emulsion has utility as an oxygen transport medium, such as artificial blood. The emulsion can optionally include addition emulsifiers of SURFYNOL®SE surfactant and PLURONIC® P-105 surfactant. The emulsion is produced using an improved emulsification technique. < 03 CM CO a. Ill Xerox Copy Centre 0 282 949 CONCENTRATED STABLE FLUOROCHEMICAL AQUEOUS EMULSIONS TECHNICAL FIELD The present invention is directed to biologically acceptable oxygen transport media comprising high s concentration aqueous emulsions of perfluorochemicals in complex emulsification systems. More specifi- cally, the present invention is directed to an aqueous perfluorochemical emulsion having utility in the field of resuscitative fluids for oxygen transport and volume expansion in mammals, such as artificial or synthetic blood. -

(Title of the Thesis)*
FLUOROCARBENE, FLUOROALKYL, AND FLUORIDE COMPLEXES OF FIRST-ROW TRANSITION METALS Graham Mark Lee Thesis submitted to the Faculty of Graduate and Postdoctoral Studies University of Ottawa In partial fulfillment of the requirements for the degree of Doctor of Philosophy Ottawa-Carleton Chemistry Institute Faculty of Science University of Ottawa © Graham Mark Lee, Ottawa, Canada, 2017 Abstract Fluorinated organic compounds play important roles in our society, as these products range from life-saving pharmaceuticals and agrochemicals, to fluoropolymers with extremely high thermal and chemical stability. Although elemental fluorine (F2) is the most reactive element, some fluoro- organic compounds are chemically inert. As such, controlled reactivity of fluorine or highly- fluorinated organic fragments is a considerable, yet important challenge for synthetic chemists. Fluoro-organometallic chemistry has been studied for decades, as researchers attempt to maximize the potential of metal mediated/catalyzed processes for the synthesis of fluorinated organic molecules. Within this framework, metal fluorocarbene complexes are particularly interesting because of their highly tunable reactivity, and are proposed for use in important metathesis/polymerization reactions of perfluorinated alkenes. While considerable work is still needed to make these proposed reactions a reality, this thesis outlines contributions from our F F research group. We showed that cobalt fluorocarbene complexes CpCo(=CFR )(PPh2Me) (R = F, CF3) undergo [2+2] cycloaddition reactions with tetrafluoroethylene (TFE) and phenylacetylene to form perfluorometallacyclobutane and partially fluorinated metallacyclobutene products, respectively. For both reactions, computational studies reveal a stepwise ring-closing mechanism, which proceeds through a singlet 1,4-diradical intermediate. Next, the formation of CpCo(=CF2)(L) complexes is achieved via the direct addition of difluorocarbene, generated in situ, to a cobalt(I) precursor. -

Elemental Fluorine Product Information (Pdf)
Elemental Fluorine Contents 1 Introduction ............................................................................................................... 4 2.1 Technical Application of Fluorine ............................................................................. 5 2.2 Electronic Application of Fluorine ........................................................................... 7 2.3 Fluorine On-Site Plant ............................................................................................ 8 3 Specifications ............................................................................................................ 9 4 Safety ...................................................................................................................... 10 4.1 Maintenance of the F2 system .............................................................................. 12 4.2 First Aid ................................................................................................................ 13 5.1 Chemical Properties ............................................................................................. 14 5.2 Physical Data ....................................................................................................... 15 6 Toxicity .................................................................................................................... 18 7 Shipping and Transport ........................................................................................... 20 8 Environment ........................................................................................................... -

SAFETY DATA SHEET Halocarbon R-503
SAFETY DATA SHEET Halocarbon R-503 Section 1. Identification GHS product identifier : Halocarbon R-503 Other means of : Not available. identification Product type : Liquefied gas Product use : Synthetic/Analytical chemistry. SDS # : 007306 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Liquefied gas substance or mixture HAZARDOUS TO THE OZONE LAYER - Category 1 GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May cause frostbite. May displace oxygen and cause rapid suffocation. Harms public health and the environment by destroying ozone in the upper atmosphere. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Always keep container in upright position. Prevention : Not applicable. Response : Not applicable. Storage : Protect from sunlight. Store in a well-ventilated place. Disposal : Refer to manufacturer or supplier for information on recovery or recycling. Hazards not otherwise : Liquid can cause burns similar to frostbite. -

(VI) and Chromium (V) Oxide Fluorides
Portland State University PDXScholar Dissertations and Theses Dissertations and Theses 1976 The chemistry of chromium (VI) and chromium (V) oxide fluorides Patrick Jay Green Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/open_access_etds Part of the Chemistry Commons Let us know how access to this document benefits ou.y Recommended Citation Green, Patrick Jay, "The chemistry of chromium (VI) and chromium (V) oxide fluorides" (1976). Dissertations and Theses. Paper 4039. https://doi.org/10.15760/etd.5923 This Thesis is brought to you for free and open access. It has been accepted for inclusion in Dissertations and Theses by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. All ABSTRACT OF THE TllESIS OF Patrick Jay Green for the Master of Science in Chemistry presented April 16, 1976. Title: Chemistry of Chromium(VI) and Chromium(V) Oxide Fluorides. APPROVEO BY MEMBERS OF THE THESIS CO'"o\l TIEE: y . • Ii . ' I : • • • • • New preparative routes to chromyl fluoride were sought. It was found that chlorine ironofluoride reacts with chromium trioxide and chromyl chlo ride to produce chromyl fluoride. Attempts were ~ade to define a mechan ism for the reaction of ClF and Cr0 in light of by-products observed 3 and previous investigations. Carbonyl fluoride and chromium trioxide react to fom chro·yl fluoride and carbo:i dioxide. A mechanism was also proposed for this react10n. Chromium trioxide 11itl\ l~F6 or WF5 reacts to produce chromyl fluoride and the respective oxide tetrafluoride. 2 Sulfur hexafluoride did not react with Cr03. -
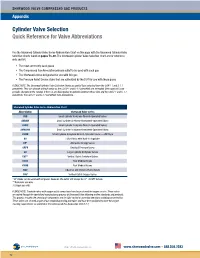
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Use of Chlorofluorocarbons in Hydrology : a Guidebook
USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A Guidebook USE OF CHLOROFLUOROCARBONS IN HYDROLOGY A GUIDEBOOK 2005 Edition The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GREECE PANAMA ALBANIA GUATEMALA PARAGUAY ALGERIA HAITI PERU ANGOLA HOLY SEE PHILIPPINES ARGENTINA HONDURAS POLAND ARMENIA HUNGARY PORTUGAL AUSTRALIA ICELAND QATAR AUSTRIA INDIA REPUBLIC OF MOLDOVA AZERBAIJAN INDONESIA ROMANIA BANGLADESH IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ SAUDI ARABIA BELGIUM IRELAND SENEGAL BENIN ISRAEL SERBIA AND MONTENEGRO BOLIVIA ITALY SEYCHELLES BOSNIA AND HERZEGOVINA JAMAICA SIERRA LEONE BOTSWANA JAPAN BRAZIL JORDAN SINGAPORE BULGARIA KAZAKHSTAN SLOVAKIA BURKINA FASO KENYA SLOVENIA CAMEROON KOREA, REPUBLIC OF SOUTH AFRICA CANADA KUWAIT SPAIN CENTRAL AFRICAN KYRGYZSTAN SRI LANKA REPUBLIC LATVIA SUDAN CHAD LEBANON SWEDEN CHILE LIBERIA SWITZERLAND CHINA LIBYAN ARAB JAMAHIRIYA SYRIAN ARAB REPUBLIC COLOMBIA LIECHTENSTEIN TAJIKISTAN COSTA RICA LITHUANIA THAILAND CÔTE D’IVOIRE LUXEMBOURG THE FORMER YUGOSLAV CROATIA MADAGASCAR REPUBLIC OF MACEDONIA CUBA MALAYSIA TUNISIA CYPRUS MALI TURKEY CZECH REPUBLIC MALTA UGANDA DEMOCRATIC REPUBLIC MARSHALL ISLANDS UKRAINE OF THE CONGO MAURITANIA UNITED ARAB EMIRATES DENMARK MAURITIUS UNITED KINGDOM OF DOMINICAN REPUBLIC MEXICO GREAT BRITAIN AND ECUADOR MONACO NORTHERN IRELAND EGYPT MONGOLIA UNITED REPUBLIC EL SALVADOR MOROCCO ERITREA MYANMAR OF TANZANIA ESTONIA NAMIBIA UNITED STATES OF AMERICA ETHIOPIA NETHERLANDS URUGUAY FINLAND NEW ZEALAND UZBEKISTAN FRANCE NICARAGUA VENEZUELA GABON NIGER VIETNAM GEORGIA NIGERIA YEMEN GERMANY NORWAY ZAMBIA GHANA PAKISTAN ZIMBABWE The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. -

Safety Data Sheet
SAFETY DATA SHEET Preparation Date: 05/19/2015 Revision Date: 05/08/2017 Revision Number: G2 1. IDENTIFICATION Product identifier Product code: B1100 Product Name: BISMUTH METAL, GRANULAR, REAGENT Other means of identification Synonyms: Bismuth-209 Bismuth, elemental CAS #: 7440-69-9 RTECS # EB2600000 CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: In safety devices in fire detection and extinguishing systems; Catalyst for making acrylic fibers; In production of malleable irons; carrier for radioactive uranium fuel in atomic reactor; In printing industry; alloying agent; chemical intermediate for pharmaceuticals and other chemicals; In cosmetics. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000. Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is not considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Not a dangerous substance or mixture according to the Globally Harmonized System (GHS) Label elements Not classified Hazards not otherwise classified (HNOC) Not Applicable Other hazards May be harmful if swallowed Product code: B1100 Product name: BISMUTH METAL, 1 / 11 GRANULAR, REAGENT 3. COMPOSITION/INFORMATION ON INGREDIENTS Components CAS-No. Weight % Bismuth Metal 7440-69-9 100 4. FIRST AID MEASURES First aid measures General Advice: National Capital Poison Center in the United States can provide assistance if you have a poison emergency and need to talk to a poison specialist. -

Periodic Trends in the Main Group Elements
Chemistry of The Main Group Elements 1. Hydrogen Hydrogen is the most abundant element in the universe, but it accounts for less than 1% (by mass) in the Earth’s crust. It is the third most abundant element in the living system. There are three naturally occurring isotopes of hydrogen: hydrogen (1H) - the most abundant isotope, deuterium (2H), and tritium 3 ( H) which is radioactive. Most of hydrogen occurs as H2O, hydrocarbon, and biological compounds. Hydrogen is a colorless gas with m.p. = -259oC (14 K) and b.p. = -253oC (20 K). Hydrogen is placed in Group 1A (1), together with alkali metals, because of its single electron in the valence shell and its common oxidation state of +1. However, it is physically and chemically different from any of the alkali metals. Hydrogen reacts with reactive metals (such as those of Group 1A and 2A) to for metal hydrides, where hydrogen is the anion with a “-1” charge. Because of this hydrogen may also be placed in Group 7A (17) together with the halogens. Like other nonmetals, hydrogen has a relatively high ionization energy (I.E. = 1311 kJ/mol), and its electronegativity is 2.1 (twice as high as those of alkali metals). Reactions of Hydrogen with Reactive Metals to form Salt like Hydrides Hydrogen reacts with reactive metals to form ionic (salt like) hydrides: 2Li(s) + H2(g) 2LiH(s); Ca(s) + H2(g) CaH2(s); The hydrides are very reactive and act as a strong base. It reacts violently with water to produce hydrogen gas: NaH(s) + H2O(l) NaOH(aq) + H2(g); It is also a strong reducing agent and is used to reduce TiCl4 to titanium metal: TiCl4(l) + 4LiH(s) Ti(s) + 4LiCl(s) + 2H2(g) Reactions of Hydrogen with Nonmetals Hydrogen reacts with nonmetals to form covalent compounds such as HF, HCl, HBr, HI, H2O, H2S, NH3, CH4, and other organic and biological compounds.