(VI) and Chromium (V) Oxide Fluorides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SULFUR TRIOXIDE -- Chemical Fact Sheet
OLEUM/SULFUR TRIOXIDE -- Chemical Fact Sheet 1 What is it? Oleum is a cloudy, gray, fuming, oily, corrosive liquid with a sharp, penetrating odor. When Oleum comes into contact with air following a spill, it releases Sulfur Trioxide. Sulfur Trioxide is a white gas having the appearance of fog. It also has a sharp, penetrating odor that is detectable at low concentrations. Because of the tendency to liberate Sulfur Trioxide on contact with air, Oleum is also known as “fuming Sulfuric Acid”. Where does it Oleum is made by dissolving Sulfur Trioxide into Sulfuric Acid. Sulfur come from? Trioxide is made from Sulfur Dioxide in the presence of a catalyst. What are the It is used in the oil refining process to make crude oil distillates into higher quality materials. common uses for it? Manufacture of soap Manufacture of high purity Sulfuric Acid for the electronic industry Manufacture of catalyst used in production of Sulfuric Acid. How is it Oleum is shipped by truck and pipeline. transported in CCC? How is it stored Oleum is stored in covered tanks. in CCC? Health Hazards from Exposure Exposure Route Symptoms First Aid Inhalation Irritates nose, throat and Remove to fresh air. Seek (low concentrations) lungs medical attention if Burning Sensation symptoms persist. Sneezing, coughing Inhalation Burning sensation Remove to fresh air, get (high concentrations & prolonged exposure) Coughing, gagging medical attention including Chest tightness and pain, oxygen administration. Fluid in lungs Initiate CPR if breathing has Suffocation, death stopped. Eyes Severely irritates eyes Rinse eyes with water for at Burning/discomfort least 5 minutes. -

Source Test Method ST-20 SULFUR DIOXIDE, SULFUR TRIOXIDE
Source Test Method ST-20 SULFUR DIOXIDE, SULFUR TRIOXIDE, SULFURIC ACID MIST (Adopted January 20, 1982) REF: Regulations 6-320, 6-330, 9-1-302, 9-1-304 thru 310, 10-1-301, 12-6-301 1. APPLICABILITY 1.1 This method is used to quantify emissions of s ulfur dioxide, sulfur trioxide and sulfuric acid mist. It determines compliance with Regulations 6-320 and 6-330 for SULFUR TRIOXIDE and SULFURIC ACID MIST, and 9-1-302, 9- 1-304 thru 310 and 10-1-301 and 12-6-301 for SULFUR DIOXIDE. 1.2 This method, modified with a glass fiber disc filter as the back-up SO 3 filter, has been given alternate status by the EPA to EPA Method 8. It may be used to determine compliance with oxides of sulfur regulations under Regulation 9. 2. PRINCIPLE 2.1 Sulfuric acid mist, sulfur trioxide and sulfur dioxide are collected in a single extractive sampling train. Acid mist is trapped in a quartz wool plug in the sample probe and is subsequently analyzed with an acid-base titration. Sulfur trioxide is absorbed in an 80% isopropyl alcohol (IPA)/water solution with a quartz wool back-up filter and is analyzed using analytical procedure Lab-12. Sulfur dioxide is absorbed in an aqueous hydrogen peroxide solution and is analyzed using analytical procedure Lab-12. 3. RANGE 3.1 The minimum measurable concentration using this method listed below are: 3 3.1.1 Acid mist - 0.0002 gr/ft as H2SO4 3.1.2 Sulfur trioxide - 7 ppm 3.1.3 Sulfur dioxide - 7 ppm 3.2 The maximum measurable concentrations using this method listed below are: 3.2.1 Acid mist - undetermined 3.2.2 Sulfur trioxide - 350 ppm 3.2.3 Sulfur dioxide - 2.5 % 4. -

Downloaded for Personal Non-Commercial Research Or Study, Without Prior Permission Or Charge
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] REDUCTION OF MOLYBDENUM AND TUNGSTEN HEXAFLUORIDES TO THE PENTAVALENT STATE. A thesis submitted to the University of Glasgow for the degree of M.Sc. in the Faculty of Science. by NASHUNMENGHE BAO Department of Chemistry, University of Glasgow. November 1989, (c) Nashunmenghe ProQuest Number: 10999292 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10999292 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. -

Packet of Wiser Reports on Acetone Acetonitrile
Ac&tone ^Hazmat - NFPA Hazard Classification Page 1 of Acetone CAS RN: 67-64-1 Hazmat - NFPA Hazard Classification SOMS DocID 2085807 Health: 1 (Slight) Materials that, on exposure, would cause significant irritation, but only minor residual injury, including those requiring the use of an approved air-purifying respirator. These materials are only slightly hazardous to health and only breathing protection is needed. Flammability: 3 (Severe) rhis degree includes Class IB and 1C flammable liquids and materials that can be easily ignited under almost all normal temperature conditions. Water may be ineffective in controlling or extinguishing fires in such materials. Instability: 0 (Minimal) This degree includes materials that are normally stable, even under fire exposure conditions, and that do not react with water.- Norma lire fighting procedures may be used. Printed by WISER for Windows (v2.3.231, database v2.108) HHS/NIH, National Library of Medicine AR000018 iile://C:\Documents and Settings\Gham\Application Data\National Library of Medicine\WISER\2.3.231.628... 9/27/20 Acetone ^Key Info Page 1 of Acetone CAS RN: 67-64-1 Key Info FLAMMABLE LIQUIDS (Polar / Water-Miscible) • HIGHLY FLAMMABLE: Easily ignited by heat, sparks or flames • CAUTION: Very low flash point; use of water spray when fighting fire may be inefficient Printed by WISER for Windows (v2.3.231, database v2.108) HHS/NIH, National Library of Medicine AR000019 file://C:\Documents and Settings\Gham\Application Data\National Library of Medicine\WISER\2.3.231.628../ 9/27/20 Acetone - -Hazmat - Explosive Limits / Potential Page 1 of Acetone CAS RIM: 67-64-1 Hazmat - Explosive Limits / Potential Highly flammable liquid. -

Wood Modified by Inorganic Salts: Mechanism and Properties
WOOD MODIFIED BY INORGANIC SALTS: MECHANISM AND PROPERTIES. I. WEATHERING RATE, WATER REPELLENCY, AND IIIMENSIONAL STABILITY OF WOOD MODIFIEID WITH CHROMIUM (111) NITRATE VERSUS CHROMIC ACID' R. S. Willianzs and W. C. Feisl Research Chemists Forest Products Lab~ratory,~Forest Service U.S. Department of Agriculture Madison, WI 53705 (Received October 1983) ABSTRACT Chromic acid treatment of wood improves surface characteristics. A simple dip or brush application of 5% aqueous chromic acid to a wood surface stops extractive staining, improves dimensional stability, retards weathering of unfinished wood, and prolongs the life of finishes. A better understanding of how chromic acid effects these improvements may facilitate the development of even better treatments. The improvements may be due to a combination offactors: oxidation ofwood by hexavalent chromium compounds, formation of coordination coml~lexeswith trivalent chromium, and the presence of insoluble chromium compounds at the surface. Most trivalent chromium compounds do not become fixed in wood (i.e., do not form water-insolublf: wood-chemical complexes). However, chromium (111) nitrate treatment of western redcedar (Thuja plicata), southern pine (Pinus sp.), and ponderosa pine (Pinus ponderosa) produced modified wood with properties similar to chromic acid-treated wood. Evaluation of the chromium (111) nitrate- and chromic acid-modified wood by leaching, Xenon arc- accelerated weathering, and swellometer experiments clearly demonstrated similar fixation, erosion, and water-repellent properties. Based on the results from chromium (111) nitrate-treated wood, fixation through coordination of trivalent chromium with wood hydroxyls an(5 formation of insoluble chromium appear to be the critical factors in improving the performance of the treated wood. -

Fluorosulfates of Silver, Ruthenium, and Osmium
FLUOROSULFATES OF SILVER, RUTHENIUM, AND OSMIUM by PATRICK CHEUNG SHING LEUNG B.Sc. (Hons.), The University of British Columbia, 1975 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES (Department of Chemistry) We accept this thesis as conforming to the required standard THE UNIVERSITY OF BRITISH COLUMBIA December, 1979 © Patrick Cheung Shing Leung, 1979 In presenting this thesis in partial fulfilment of the requirements f an advanced degree at the University of British Columbia, I agree tha the Library shall make it freely available for reference and study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by his representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of CMMUTtY The University of British Columbia 2075 Wesbrook Place Vancouver, Canada V6T 1W5 ABSTRACT A number of synthetic routes to silver(II) fluorosulfate, Ag(SC>3F)2, were systematically explored. The most suitable and versatile route was found to be the oxidation of silver metal by a solution of bisfluorosulfuryl peroxide, S20gF2, in fluorosulfuric acid, HSO^F, according to: HS03F Ag + S206F2 m- Ag (SC>3F) 2> Additional methods which were found to be suitable involved the oxidation of a wide variety of silver(I) compounds such as S F or the nsert:LOn of S0 Ag20 or AgSO^F by 2°6 2' i- 3 into AgF2. Structural conclusions on Ag(S03F)2 and the other compounds synthesized subsequently were based on the vibrational, elec• tronic and electron spin resonance spectra, as well as on magnetic susceptibility measurements made between 300 and 77 K. -

Chromium Compounds Hazard Summary
Chromium Compounds Hazard Summary Chromium occurs in the environment primarily in two valence states, trivalent chromium (Cr III) and hexavalent chromium (Cr VI). Exposure may occur from natural or industrial sources of chromium. Chromium III is much less toxic than chromium (VI). The respiratory tract is also the major target organ for chromium (III) toxicity, similar to chromium (VI). Chromium (III) is an essential element in humans. The body can detoxify some amount of chromium (VI) to chromium (III). The respiratory tract is the major target organ for chromium (VI) toxicity, for acute (short-term) and chronic (long-term) inhalation exposures. Shortness of breath, coughing, and wheezing were reported from a case of acute exposure to chromium (VI), while perforations and ulcerations of the septum, bronchitis, decreased pulmonary function, pneumonia, and other respiratory effects have been noted from chronic exposure. Human studies have clearly established that inhaled chromium (VI) is a human carcinogen, resulting in an increased risk of lung cancer. Animal studies have shown chromium (VI) to cause lung tumors via inhalation exposure. Please Note: The main sources of information for this fact sheet are EPA's Integrated Risk Information System (IRIS) (7), which contains information on inhalation chronic toxicity and the RfC and oral chronic toxicity and the RfD, and the carcinogenic effects of chromium including the unit cancer risk for inhalation exposure, EPA's Toxicological Review of Trivalent Chromium and Toxicological Review of Hexavalent Chromium (3), and the Agency for Toxic Substances and Disease Registry's (ATSDR's) Toxicological Profile for Chromium. (1) Uses The metal chromium is used mainly for making steel and other alloys. -

Chlorine Trifluoride
MATERIAL SAFETY DATA SHEET Prepared to U.S. OSHA, CMA, ANSI and Canadian WHMIS Standards 1. PRODUCT IDENTIFICATION CHEMICAL NAME; CLASS: CHLORINE TRIFLUORIDE SYNONYMS: Chlorine Fluoride CHEMICAL FAMILY NAME: Halogen Fluoride FORMULA: ClF3 Document Number: 20026 PRODUCT USE: Use as a fluorinator; for cutting oil-well tubes; reprocessing reactor fuels, as an oxidizer in propellants. SUPPLIER/MANUFACTURER'S NAME: AIR LIQUIDE AMERICA CORPORATION ADDRESS: 2700 Post Oak Drive Houston, TX 77056-8229 EMERGENCY PHONE: CHEMTREC: 1-800-424-9300 BUSINESS PHONE: General MSDS Information 1-713/896-2896 Fax on Demand: 1-800/231-1366 2. COMPOSITION and INFORMATION ON INGREDIENTS CHEMICAL NAME CAS # mole % EXPOSURE LIMITS IN AIR ACGIH OSHA TLV STEL PEL STEL IDLH OTHER ppm ppm ppm ppm ppm Chlorine Trifluoride 7790-91-2 > 99% NE 0.1, C NE 0.1, C 20 NIOSH REL: 0.1 C ppm DFG MAK: 0.1 ppm, C Maximum Impurities < 1% None of the trace impurities in this product contribute significantly to the hazards associated with the product. All hazard information pertinent to this product has been provided in this Material Safety Data Sheet, per the requirements of the OSHA Hazard Communication Standard (29 CFR 1910.1200) and State equivalents standards. NE = Not Established C = Ceiling Limit See Section 16 for Definitions of Terms Used. NOTE: all WHMIS required information is included. It is located in appropriate sections based on the ANSI Z400.1-1993 format. CHLORINE TRIFLUORIDE - ClF3 MSDS EFFECTIVE DATE: JUNE 1, 1998 PAGE 1 OF 9 3. HAZARD IDENTIFICATION EMERGENCY OVERVIEW: Chlorine Trifluoride is an extremely toxic, corrosive, water-reactive, oxidizing, colorless, liquefied gas, with a suffocating, sweet odor. -

Safety Data Sheet
SAFETY DATA SHEET Preparation Date: 05/19/2015 Revision Date: 05/08/2017 Revision Number: G2 1. IDENTIFICATION Product identifier Product code: B1100 Product Name: BISMUTH METAL, GRANULAR, REAGENT Other means of identification Synonyms: Bismuth-209 Bismuth, elemental CAS #: 7440-69-9 RTECS # EB2600000 CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: In safety devices in fire detection and extinguishing systems; Catalyst for making acrylic fibers; In production of malleable irons; carrier for radioactive uranium fuel in atomic reactor; In printing industry; alloying agent; chemical intermediate for pharmaceuticals and other chemicals; In cosmetics. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000. Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is not considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Not a dangerous substance or mixture according to the Globally Harmonized System (GHS) Label elements Not classified Hazards not otherwise classified (HNOC) Not Applicable Other hazards May be harmful if swallowed Product code: B1100 Product name: BISMUTH METAL, 1 / 11 GRANULAR, REAGENT 3. COMPOSITION/INFORMATION ON INGREDIENTS Components CAS-No. Weight % Bismuth Metal 7440-69-9 100 4. FIRST AID MEASURES First aid measures General Advice: National Capital Poison Center in the United States can provide assistance if you have a poison emergency and need to talk to a poison specialist. -

United States Patent Office Patented Dec
3,629,301 United States Patent Office Patented Dec. 21, 1971 3,629,301 3,3-DIFLUORO-2-SUBSTITUTED STEROIDS AND THER PREPARATION William C. Ripka, Wilmington, Del, assignor to E. I. du Pont de Nemours and Company, Wilmington, Del. 5 where R is hydrogen, methyl, or ethynyl, and R2 is hydro No Drawing. Filed Oct. 24, 1969, Ser. No. 869,352 gen or an alkanoyl having no more than 8 carbon atoms, Int, C. C07c 169/20 Such as formyl, acetyl, propionyl, butyryl, valleryl and U.S. C. 260-397.3 10 Claims caproyl. These compounds are prepared from 3-fluoro-A2-steroids 10 by a reaction with nitrosyl fluoride and subsequent conver ABSTRACT OF THE DISCLOSURE sion of the 3,3-difluoro-2-nitrimino steroid products to the New steroids of the formula corresponding 3,3-difluoro-2-keto steroids with alumina CE containing Water, as shown in the reaction scheme below X involving the steroid A-ring: R NO R R ? NOF N= A. AlO3.H2O Os R - A. l -3 F2 Fs! N Z-F 20 The 2-keto group can be converted to the 2-hydroxy F= group by conventional techniques, including, for example, wherein Z is oxygen or reduction with sodium borohydrides. DETALED DESCRIPTION OF THE INVENTION The first step of the reaction sequence, the reaction of a .O 3-fluoro-Asteroid with nitrosyl fluoride is carried out in R is hydrogen or methyl; and X is oxygen or an inert Solvent such as methylene chloride, chloroform, OR2 carbon tetrachloride, fluorodichloromethane, and ethylene 30 dichloride. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -
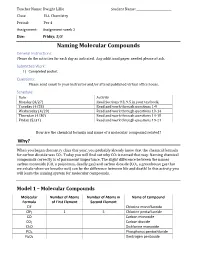
Naming Molecular Compounds General Instructions: Please Do the Activities for Each Day As Indicated
Teacher Name: Dwight Lillie Student Name: ________________________ Class: ELL Chemistry Period: Per 4 Assignment: Assignment week 2 Due: Friday, 5/8 Naming Molecular Compounds General Instructions: Please do the activities for each day as indicated. Any additional paper needed please attach. Submitted Work: 1) Completed packet. Questions: Please send email to your instructor and/or attend published virtual office hours. Schedule: Date Activity Monday (4/27) Read Sections 9.3, 9.5 in your textbook. Tuesday (4/28) Read and work through questions 1-9 Wednesday (4/29) Read and work through questions 10-14 Thursday (4/30) Read and work through questions 14-18 Friday (5/31) Read and work through questions 19-21 How are the chemical formula and name of a molecular compound related? Why? When you began chemistry class this year, you probably already knew that the chemical formula for carbon dioxide was CO2. Today you will find out why CO2 is named that way. Naming chemical compounds correctly is of paramount importance. The slight difference between the names carbon monoxide (CO, a poisonous, deadly gas) and carbon dioxide (CO2, a greenhouse gas that we exhale when we breathe out) can be the difference between life and death! In this activity you will learn the naming system for molecular compounds. Model 1 – Molecular Compounds Molecular Number of Atoms Number of Atoms in Name of Compound Formula of First Element Second Element ClF Chlorine monofluoride ClF5 1 5 Chlorine pentafluoride CO Carbon monoxide CO2 Carbon dioxide Cl2O Dichlorine monoxide PCl5 Phosphorus pentachloride N2O5 Dinitrogen pentoxide 1. Fill in the table to indicate the number of atoms of each type in the molecular formula.