A Molecular Model for Neurodevelopmental Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -

Understanding the Impact of 1Q21.1 Copy Number Variant
Understanding the impact of 1q21.1 copy number variant Article (Published Version) Havard, Chansonette, Strong, Emma, Mercier, Eloi, Colnaghi, Rita, Alcantara, Diana Rita Ralha, Chow, Eva, Martell, Sally, Tyson, Christine, Hrynchak, Monica, McGillivray, Barbara, Hamilton, Sara, Marles, Sandra, Mhanni, Aziz, Dawson, Angelika J, Pavlidis, Paul et al. (2011) Understanding the impact of 1q21.1 copy number variant. Orphanet Journal of Rare Diseases, 6 (54). pp. 1-12. ISSN 1750-1172 This version is available from Sussex Research Online: http://sro.sussex.ac.uk/id/eprint/41577/ This document is made available in accordance with publisher policies and may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the URL above for details on accessing the published version. Copyright and reuse: Sussex Research Online is a digital repository of the research output of the University. Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. To the extent reasonable and practicable, the material made available in SRO has been checked for eligibility before being made available. Copies of full text items generally can be reproduced, displayed or performed and given to third parties in any format or medium for personal research or study, educational, or not-for-profit purposes without prior permission or charge, provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. -
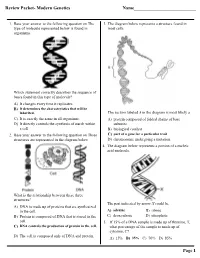
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Statistical Analysis of the Genomic Distribution and Correlation of Regulatory Elements in the ENCODE Regions
Downloaded from genome.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Statistical analysis of the genomic distribution and correlation of regulatory elements in the ENCODE regions Zhengdong D. Zhang,1 Alberto Paccanaro,2 Yutao Fu,3 Sherman Weissman,5 Zhiping Weng,3,4 Joseph Chang,6 Michael Snyder,7 and Mark B. Gerstein1,8,9 1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06520, USA; 2Department of Computer Science Royal Holloway, University of London, Egham Hill, TW20 0EX, United Kingdom; 3Bioinformatics Program, Boston University, Boston, Massachusetts 02215, USA; 4Biomedical Engineering Department, Boston University, Boston, Massachusetts 02215, USA; 5Department of Genetics, Yale University, New Haven, Connecticut 06510, USA; 6Department of Statistics, Yale University, New Haven, Connecticut 06520, USA; 7Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, Connecticut 06520, USA; 8Program in Computational Biology and Bioinformatics Yale University, New Haven, Connecticut 06520, USA The comprehensive inventory of functional elements in 44 human genomic regions carried out by the ENCODE Project Consortium enables for the first time a global analysis of the genomic distribution of transcriptional regulatory elements. In this study we developed an intuitive and yet powerful approach to analyze the distribution of regulatory elements found in many different ChIP–chip experiments on a 10∼100-kb scale. First, we focus on the overall chromosomal distribution of regulatory elements in the ENCODE regions and show that it is highly nonuniform. We demonstrate, in fact, that regulatory elements are associated with the location of known genes. Further examination on a local, single-gene scale shows an enrichment of regulatory elements near both transcription start and end sites. -

Application of X-Ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-19-2008 Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis Zakhia Moore University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Moore, Zakhia, "Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis" (2008). University of New Orleans Theses and Dissertations. 888. https://scholarworks.uno.edu/td/888 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Chemistry by Zakhia Moore B.S., Xavier University, 2000 M.S., University of New Orleans, 2005 December, 2008 ACKNOWLEDGEMENTS Giving all glory to God for His many blessings, whom without I could not have completed this long journey. -

Genetic Screens in Isogenic Mammalian Cell Lines Without Single Cell Cloning
ARTICLE https://doi.org/10.1038/s41467-020-14620-6 OPEN Genetic screens in isogenic mammalian cell lines without single cell cloning Peter C. DeWeirdt1,2, Annabel K. Sangree1,2, Ruth E. Hanna1,2, Kendall R. Sanson1,2, Mudra Hegde 1, Christine Strand 1, Nicole S. Persky1 & John G. Doench 1* Isogenic pairs of cell lines, which differ by a single genetic modification, are powerful tools for understanding gene function. Generating such pairs of mammalian cells, however, is labor- 1234567890():,; intensive, time-consuming, and, in some cell types, essentially impossible. Here, we present an approach to create isogenic pairs of cells that avoids single cell cloning, and screen these pairs with genome-wide CRISPR-Cas9 libraries to generate genetic interaction maps. We query the anti-apoptotic genes BCL2L1 and MCL1, and the DNA damage repair gene PARP1, identifying both expected and uncharacterized buffering and synthetic lethal interactions. Additionally, we compare acute CRISPR-based knockout, single cell clones, and small- molecule inhibition. We observe that, while the approaches provide largely overlapping information, differences emerge, highlighting an important consideration when employing genetic screens to identify and characterize potential drug targets. We anticipate that this methodology will be broadly useful to comprehensively study gene function across many contexts. 1 Genetic Perturbation Platform, Broad Institute of MIT and Harvard, 75 Ames Street, Cambridge, MA 02142, USA. 2These authors contributed equally: Peter C. DeWeirdt, -

Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Defi Cient Prostate Cancer
Published OnlineFirst May 8, 2020; DOI: 10.1158/2159-8290.CD-19-1352 RESEARCH ARTICLE Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Defi cient Prostate Cancer Di Zhao 1 , 2 , Li Cai 1 , Xin Lu 1 , 3 , Xin Liang 1 , 4 , Jiexi Li 1 , Peiwen Chen 1 , Michael Ittmann 5 , Xiaoying Shang 1 , Shan Jiang6 , Haoyan Li 2 , Chenling Meng 2 , Ivonne Flores 6 , Jian H. Song 4 , James W. Horner 6 , Zhengdao Lan 1 , Chang-Jiun Wu6 , Jun Li 6 , Qing Chang 7 , Ko-Chien Chen 1 , Guocan Wang 1 , 4 , Pingna Deng 1 , Denise J. Spring 1 , Y. Alan Wang1 , and Ronald A. DePinho 1 Downloaded from cancerdiscovery.aacrjournals.org on September 28, 2021. © 2020 American Association for Cancer Research. Published OnlineFirst May 8, 2020; DOI: 10.1158/2159-8290.CD-19-1352 ABSTRACT Genetic inactivation of PTEN is common in prostate cancer and correlates with poorer prognosis. We previously identifi edCHD1 as an essential gene in PTEN- defi cient cancer cells. Here, we sought defi nitivein vivo genetic evidence for, and mechanistic under- standing of, the essential role of CHD1 in PTEN-defi cient prostate cancer. In Pten and Pten /Smad4 genetically engineered mouse models, prostate-specifi c deletion ofChd1 resulted in markedly delayed tumor progression and prolonged survival. Chd1 deletion was associated with profound tumor microenvironment (TME) remodeling characterized by reduced myeloid-derived suppressor cells (MDSC) and increased CD8+ T cells. Further analysis identifi ed IL6 as a key transcriptional target of CHD1, which plays a major role in recruitment of immunosuppressive MDSCs. -

1Q21.1 Duplication Syndrome and Epilepsy Case Report and Review
CLINICAL/SCIENTIFIC NOTES OPEN ACCESS 1q21.1 Duplication syndrome and epilepsy Case report and review Ioulia Gourari, MD, Romaine Schubert, MD, and Aparna Prasad, PhD Correspondence Dr. Gourari Neurol Genet 2018;4:e219. doi:10.1212/NXG.0000000000000219 [email protected] Copy number variants (CNVs) of 1q21.1 are increasingly being recognized due to the wide- spread use of genetic screening tests for the investigation of developmental disorders and epilepsy. These include microdeletion and microduplication syndromes, associated with a wide variety of pathology including autism spectrum disorders, attention-deficit disorder, learning disabilities, hypotonia, facial dysmorphisms, and schizophrenia. The 1q21.1 region is consid- ered to be genetically unstable because it contains one of the largest areas of identical dupli- cation sequences in the human genome. Epilepsy has been reported in the literature, particularly in microdeletion syndromes, but rarely in association with microduplication syn- dromes. We report a patient with epilepsy and autism spectrum disorder due to a distal 1q21.1 microduplication and review the available literature and genetic information. Case report We present a 10-year-old girl with a low-functioning autism spectrum disorder and focal motor epilepsy. On examination, she has hypertelorism, minimal communicative language skills, and severe macrocephaly (HC = 57 cm, 3.6 SD > 99%). Seizures started at 7 years of age and consisted of head deviation to the left, generalized stiffening, clonic activity of the mouth, and fluttering of the eyelids, lasting for 1–2 minutes. Multiple video EEG recordings showed a right temporal focus with a less active, independent left temporal focus. 3T MRI scan of the brain was normal. -

Sox2-RNA Mechanisms of Chromosome Topological Control in Developing Forebrain
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.22.307215; this version posted September 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Sox2-RNA mechanisms of chromosome topological control in developing forebrain Ivelisse Cajigas1, Abhijit Chakraborty2, Madison Lynam1, Kelsey R Swyter1, Monique Bastidas1, Linden Collens1, Hao Luo1, Ferhat Ay2,3, Jhumku D. Kohtz1,4 1Department of Pediatrics, Northwestern University, Feinberg School of Medicine, Department of Human Molecular Genetics, Stanley Manne Children's Research Institute 2430 N Halsted, Chicago, IL 60614 2Centers for Autoimmunity and Cancer Immunotherapy, La Jolla Institute for Immunology, 9420 Athena Circle, La Jolla, CA, 92037, USA 3School of Medicine, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA 4To whom correspondence should be addressed: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.09.22.307215; this version posted September 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Precise regulation of gene expression networks requires the selective targeting of DNA enhancers. The Evf2 long non-coding RNA regulates Dlx5/6 ultraconserved enhancer(UCE) interactions with long-range target genes, controlling gene expression over a 27Mb region in mouse developing forebrain. Here, we show that Evf2 long range gene repression occurs through multi-step mechanisms involving the transcription factor Sox2, a component of the Evf2 ribonucleoprotein complex (RNP). -

Ingenuity Pathway Analysis of Differentially Expressed Genes Involved in Signaling Pathways and Molecular Networks in Rhoe Gene‑Edited Cardiomyocytes
INTERNATIONAL JOURNAL OF MOleCular meDICine 46: 1225-1238, 2020 Ingenuity pathway analysis of differentially expressed genes involved in signaling pathways and molecular networks in RhoE gene‑edited cardiomyocytes ZHONGMING SHAO1*, KEKE WANG1*, SHUYA ZHANG2, JIANLING YUAN1, XIAOMING LIAO1, CAIXIA WU1, YUAN ZOU1, YANPING HA1, ZHIHUA SHEN1, JUNLI GUO2 and WEI JIE1,2 1Department of Pathology, School of Basic Medicine Sciences, Guangdong Medical University, Zhanjiang, Guangdong 524023; 2Hainan Provincial Key Laboratory for Tropical Cardiovascular Diseases Research and Key Laboratory of Emergency and Trauma of Ministry of Education, Institute of Cardiovascular Research of The First Affiliated Hospital, Hainan Medical University, Haikou, Hainan 571199, P.R. China Received January 7, 2020; Accepted May 20, 2020 DOI: 10.3892/ijmm.2020.4661 Abstract. RhoE/Rnd3 is an atypical member of the Rho super- injury and abnormalities, cell‑to‑cell signaling and interaction, family of proteins, However, the global biological function and molecular transport. In addition, 885 upstream regulators profile of this protein remains unsolved. In the present study, a were enriched, including 59 molecules that were predicated RhoE‑knockout H9C2 cardiomyocyte cell line was established to be strongly activated (Z‑score >2) and 60 molecules that using CRISPR/Cas9 technology, following which differentially were predicated to be significantly inhibited (Z‑scores <‑2). In expressed genes (DEGs) between the knockout and wild‑type particular, 33 regulatory effects and 25 networks were revealed cell lines were screened using whole genome expression gene to be associated with the DEGs. Among them, the most signifi- chips. A total of 829 DEGs, including 417 upregulated and cant regulatory effects were ‘adhesion of endothelial cells’ and 412 downregulated, were identified using the threshold of ‘recruitment of myeloid cells’ and the top network was ‘neuro- fold changes ≥1.2 and P<0.05. -

Structural Basis of ALC1/CHD1L Autoinhibition and the Mechanism of Activation By
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.24.449738; this version posted June 24, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Structural basis of ALC1/CHD1L autoinhibition and the mechanism of activation by the nucleosome Li Wang,1,2,† Kangjing Chen,1,2,† and Zhucheng Chen1,2,3, * 1Key Laboratory for Protein Sciences of Ministry of Education, School of Life Science, Tsinghua University, Beijing, 100084, P.R. China 2Beijing Advanced Innovation Center for Structural Biology & Beijing Frontier Research Center for Biological Structure, School of Life Science, Tsinghua University, Beijing 100084, China 3Tsinghua-Peking Center for Life Sciences, Beijing 100084, China † These authors contributed equally: Li Wang, Kangjing Chen *Correspondence to: Zhucheng Chen Email: [email protected] Telephone: 86-10-62796096 Abstract Chromatin remodeler ALC1 (amplification in liver cancer 1) is crucial for repairing damaged DNA. It is autoinhibited and activated by nucleosomal epitopes. However, the mechanisms by which ALC1 is regulated remain unclear. Here we report the crystal structure of human ALC1 and the cryoEM structure bound to the nucleosome. The structure shows the macro domain of ALC1 binds to lobe 2 of the ATPase motor, sequestering two elements for nucleosome recognition, explaining the autoinhibition mechanism of the enzyme. The H4 tail competes with the macro domain for lobe 2- binding, explaining the requirement for this nucleosomal epitope for ALC1 activation. -

IAPS Act 15-17 Section Preview of the Teachers Edition.Pdf
Section Preview of the Teacher’s Edition for The Chemistry of Materials, Issues and Physical Science, 2nd Edition Activities 15-17 Suggested student responses and answer keys have been blocked out so that web-savvy students do not find this page and have access to answers. To experience a complete activity please request a sample found in the footer at lab-aids.com Families of Elements 15 4 s 0 2 n - o t si o es 50 te s -minu N O I T I N A ACTIVITY OVERVIEW V E S T I G Students compare physical and chemical properties of 13 elements and sort them into groups based on common properties. They then compare their classifications with groups—or families—of elements as defined by scientists and displayed in the Periodic Table of the Elements. KEY CONCEPTS AND PROCESS SKILLS (with correlation to NSE 5–8 Content Standards) 1. Elements have characteristic physical and chemical properties. Substances often are placed in categories or groups if they react in similar ways; metals are an example of such a group. (PhysSci: 1) 2. Scientists classify elements into families, based on their properties. (PhysSci: 1) 3. Students develop descriptions based on evidence. (Inquiry: 1) KEY VOCABULARY atom atomic mass element family (of elements) metal Periodic Table of the Elements B-39 Activity 15 • Families of Elements MATERIALS AND ADVANCE PREPARATION For the teacher aluminum, iron, copper, and carbon samples from Activity 14, “Physical and Chemical Properties of Materials” 1 Transparency 15.1, “Information on Element Cards” 8 sets of 4 Element Family Cards