Molecular Modelling for Beginners
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -
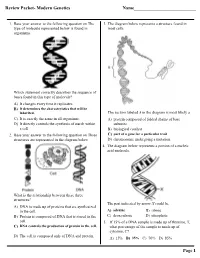
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Application of X-Ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-19-2008 Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis Zakhia Moore University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Moore, Zakhia, "Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis" (2008). University of New Orleans Theses and Dissertations. 888. https://scholarworks.uno.edu/td/888 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Application of X-ray Diffraction Methods and Molecular Mechanics Simulations to Structure Determination and Cotton Fiber Analysis A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Chemistry by Zakhia Moore B.S., Xavier University, 2000 M.S., University of New Orleans, 2005 December, 2008 ACKNOWLEDGEMENTS Giving all glory to God for His many blessings, whom without I could not have completed this long journey. -

Introduction Physics Engines AGEIA Physx SDK AGEIA
25/3/2008 SVR2007 Introduction Tutorial AGEIA PhysX • What does physics give? – Real world interaction metaphor – Realistic simulation Virtual Reality • Who needs physics? and Multimedia { mouse, ds2, gsm, masb, vt, jk } @cin.ufpe.br Research Group Thiago Souto Maior – Immersive games and applications Daliton Silva Guilherme Moura Márcio Bueno • How to use this? Veronica Teichrieb Judith Kelner – Through physics engines implemented in software or hardware Petrópolis, May 2007 Physics Engines AGEIA PhysX SDK • Simplified Newtonian models • Based upon NovodeX API • “High precision” versus “real time” simulations • Commercial and open source engines • Middleware concept • What comes together in a physics engine? • First asynchronous physics API – Mandatory • Tons of math calculations • Takes advantage of multiprocessing game • Collision detection • Rigid body dynamics systems – Optional • Fluid dynamics • Cloth simulation • Particle systems • Deformable object dynamics AGEIA PhysX SDK AGEIA PhysX SDK • Supports game • Integration with game development life cycle engines – 3DSMax and Maya – Unreal Engine 3.0 plugins (PhysX – Emergent GameBryo Create) – Natural Motion – Properties tuning with Endorphin PhysX Rocket • Runs in PC, Xbox 360 – Visual remote and PS3 debugging with PhysX VRD 1 25/3/2008 AGEIA PhysX SDK AGEIA PhysX Processor • Supports • AGEIA PhysX PPU – Complex rigid body dynamics • Powerful PPU + – Collision detection CPU + GPU – Joints and springs – Physics + Math + – Volumetric fluid simulation Fast Rendering – Particle systems -

Quantum Theory of the Hydrogen Atom
Quantum Theory of the Hydrogen Atom Chemistry 35 Fall 2000 Balmer and the Hydrogen Spectrum n 1885: Johann Balmer, a Swiss schoolteacher, empirically deduced a formula which predicted the wavelengths of emission for Hydrogen: l (in Å) = 3645.6 x n2 for n = 3, 4, 5, 6 n2 -4 •Predicts the wavelengths of the 4 visible emission lines from Hydrogen (which are called the Balmer Series) •Implies that there is some underlying order in the atom that results in this deceptively simple equation. 2 1 The Bohr Atom n 1913: Niels Bohr uses quantum theory to explain the origin of the line spectrum of hydrogen 1. The electron in a hydrogen atom can exist only in discrete orbits 2. The orbits are circular paths about the nucleus at varying radii 3. Each orbit corresponds to a particular energy 4. Orbit energies increase with increasing radii 5. The lowest energy orbit is called the ground state 6. After absorbing energy, the e- jumps to a higher energy orbit (an excited state) 7. When the e- drops down to a lower energy orbit, the energy lost can be given off as a quantum of light 8. The energy of the photon emitted is equal to the difference in energies of the two orbits involved 3 Mohr Bohr n Mathematically, Bohr equated the two forces acting on the orbiting electron: coulombic attraction = centrifugal accelleration 2 2 2 -(Z/4peo)(e /r ) = m(v /r) n Rearranging and making the wild assumption: mvr = n(h/2p) n e- angular momentum can only have certain quantified values in whole multiples of h/2p 4 2 Hydrogen Energy Levels n Based on this model, Bohr arrived at a simple equation to calculate the electron energy levels in hydrogen: 2 En = -RH(1/n ) for n = 1, 2, 3, 4, . -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Simplified Hartree-Fock Computations on Second-Row Atoms
Simplified Hartree-Fock Computations on Second-Row Atoms S. M. Blinder Wolfram Research, Inc. Champaign, IL 61820, USA May 18, 2021 Modern computational quantum chemistry has developed to a large extent beginning with applications of the Hartree-Fock method to atoms and molecules[1]. Density-functional methods have now largely supplanted conventional Hartree-Fock computations in current applications of computational chemistry. Still, from a pedagogical point of view, an un- derstanding of the original methods remains a necessary preliminary. However, since more elaborate Hartree-Fock computations are now no longer needed, it will suffice to consider a computationally simplified application of the method[2]. Accordingly, we will represent the many-electron wavefunction by a single closed-shell Slater determinant and use basis functions which are simple Slater-type orbitals. This is in contrast to the more elaborate Hartree-Fock computations, which might use multi-determinant wavefunctions and double- zeta basis functions. All of the symbolic and numerical operations were carried out using Mathematica. We consider Hartree-Fock computations on the ground states of the second-row atoms He through Ne, Z = 2 to 10, using the simplest set of orthonormalized 1s, 2s and 2p orbital functions: s α3=2 3β5 α + β = p e−αr; = 1 − r e−βr; 1s π 2s π(α2 − αβ + β2) 3 5=2 γ −γr 2pfx;y;zg = p re fsin θ cos φ, sin θ sin φ, cos θg: (1) arXiv:2105.07018v1 [quant-ph] 14 May 2021 π An orbital function multiplied by a spin function α or β is known as a spinorbital, a product of the form ( α φ(x) = (r) : (2) β 1 A simple representation of a many-electron atom is given by a Slater determinant con- structed from N occupied spin-orbitals: φa(1) φb(1) : : : φn(1) 1 φa(2) φb(2) : : : φn(2) Ψ(1;:::;N) = p ; (3) . -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

A Self-Interaction-Free Local Hybrid Functional: Accurate Binding
A self-interaction-free local hybrid functional: accurate binding energies vis-`a-vis accurate ionization potentials from Kohn-Sham eigenvalues Tobias Schmidt,1, ∗ Eli Kraisler,2, ∗ Adi Makmal,2, † Leeor Kronik,2 and Stephan K¨ummel1 1Theoretical Physics IV, University of Bayreuth, 95440 Bayreuth, Germany 2Department of Materials and Interfaces, Weizmann Institute of Science, Rehovoth 76100,Israel (Dated: September 12, 2018) We present and test a new approximation for the exchange-correlation (xc) energy of Kohn- Sham density functional theory. It combines exact exchange with a compatible non-local correlation functional. The functional is by construction free of one-electron self-interaction, respects constraints derived from uniform coordinate scaling, and has the correct asymptotic behavior of the xc energy density. It contains one parameter that is not determined ab initio. We investigate whether it is possible to construct a functional that yields accurate binding energies and affords other advantages, specifically Kohn-Sham eigenvalues that reliably reflect ionization potentials. Tests for a set of atoms and small molecules show that within our local-hybrid form accurate binding energies can be achieved by proper optimization of the free parameter in our functional, along with an improvement in dissociation energy curves and in Kohn-Sham eigenvalues. However, the correspondence of the latter to experimental ionization potentials is not yet satisfactory, and if we choose to optimize their prediction, a rather different value of the functional’s parameter is obtained. We put this finding in a larger context by discussing similar observations for other functionals and possible directions for further functional development that our findings suggest. -

Spoken Tutorial Project, IIT Bombay Brochure for Chemistry Department
Spoken Tutorial Project, IIT Bombay Brochure for Chemistry Department Name of FOSS Applications Employability GChemPaint GChemPaint is an editor for 2Dchem- GChemPaint is currently being developed ical structures with a multiple docu- as part of The Chemistry Development ment interface. Kit, and a Standard Widget Tool kit- based GChemPaint application is being developed, as part of Bioclipse. Jmol Jmol applet is used to explore the Jmol is a free, open source molecule viewer structure of molecules. Jmol applet is for students, educators, and researchers used to depict X-ray structures in chemistry and biochemistry. It is cross- platform, running on Windows, Mac OS X, and Linux/Unix systems. For PG Students LaTeX Document markup language and Value addition to academic Skills set. preparation system for Tex typesetting Essential for International paper presentation and scientific journals. For PG student for their project work Scilab Scientific Computation package for Value addition in technical problem numerical computations solving via use of computational methods for engineering problems, Applicable in Chemical, ECE, Electrical, Electronics, Civil, Mechanical, Mathematics etc. For PG student who are taking Physical Chemistry Avogadro Avogadro is a free and open source, Research and Development in Chemistry, advanced molecule editor and Pharmacist and University lecturers. visualizer designed for cross-platform use in computational chemistry, molecular modeling, material science, bioinformatics, etc. Spoken Tutorial Project, IIT Bombay Brochure for Commerce and Commerce IT Name of FOSS Applications / Employability LibreOffice – Writer, Calc, Writing letters, documents, creating spreadsheets, tables, Impress making presentations, desktop publishing LibreOffice – Base, Draw, Managing databases, Drawing, doing simple Mathematical Math operations For Commerce IT Students Drupal Drupal is a free and open source content management system (CMS). -

Rydberg Constant and Emission Spectra of Gases
Page 1 of 10 Rydberg constant and emission spectra of gases ONE WEIGHT RECOMMENDED READINGS 1. R. Harris. Modern Physics, 2nd Ed. (2008). Sections 4.6, 7.3, 8.9. 2. Atomic Spectra line database https://physics.nist.gov/PhysRefData/ASD/lines_form.html OBJECTIVE - Calibrating a prism spectrometer to convert the scale readings in wavelengths of the emission spectral lines. - Identifying an "unknown" gas by measuring its spectral lines wavelengths. - Calculating the Rydberg constant RH. - Finding a separation of spectral lines in the yellow doublet of the sodium lamp spectrum. INSTRUCTOR’S EXPECTATIONS In the lab report it is expected to find the following parts: - Brief overview of the Bohr’s theory of hydrogen atom and main restrictions on its application. - Description of the setup including its main parts and their functions. - Description of the experiment procedure. - Table with readings of the vernier scale of the spectrometer and corresponding wavelengths of spectral lines of hydrogen and helium. - Calibration line for the function “wavelength vs reading” with explanation of the fitting procedure and values of the parameters of the fit with their uncertainties. - Calculated Rydberg constant with its uncertainty. - Description of the procedure of identification of the unknown gas and statement about the gas. - Calculating resolution of the spectrometer with the yellow doublet of sodium spectrum. INTRODUCTION In this experiment, linear emission spectra of discharge tubes are studied. The discharge tube is an evacuated glass tube filled with a gas or a vapor. There are two conductors – anode and cathode - soldered in the ends of the tube and connected to a high-voltage power source outside the tube. -

Account 0103 - 5053 $6.00+0.00Ac Carbocations on Zeolites
J. Braz. Chem. Soc., Vol. 22, No. 7, 1197-1205, 2011. Printed in Brazil - ©2011 Sociedade Brasileira de Química Account 0103 - 5053 $6.00+0.00Ac Carbocations on Zeolites. Quo Vadis? Claudio J. A. Mota* and Nilton Rosenbach Jr.# Instituto de Química and INCT de Energia e Ambiente, Universidade Federal do Rio de Janeiro, Cidade Universitária, CT Bloco A, 21941-909 Rio de Janeiro-RJ, Brazil A natureza dos carbocátions adsorvidos na superfície da zeólita é discutida neste artigo, destacando estudos experimentais e teóricos. A adsorção de halogenetos de alquila sobre zeólitas trocadas com metais tem sido usada para estudar o equilíbrio entre alcóxidos, que são espécies covalentes, e carbocátions, que têm natureza iônica. Cálculos teóricos indicam que os carbocátions são mínimos (intermediários) na superfície de energia potencial e estabilizados por ligações hidrogênio com os átomos de oxigênio da estrutura zeolítica. Os resultados indicam que zeólitas se comportam como solventes sólidos, estabilizando a formação de espécies iônicas. The nature of carbocations on the zeolite surface is discussed in this account, highlighting experimental and theoretical studies. The adsorption of alkylhalides over metal-exchanged zeolites has been used to study the equilibrium between covalent alkoxides and ionic carbocations. Theoretical calculations indicated that the carbocations are minima (intermediates) on the potential energy surface and stabilized by hydrogen bonds with the framework oxygen atoms. The results indicate that zeolites behave like solid solvents, stabilizing the formation of ionic species. Keywords: zeolites, carbocations, alkoxides, catalysis, acidity 1. Introduction Thus, NH4Y can be prepared by exchange of the NaY with ammonium salt solutions. An acidic material can Zeolites are crystalline aluminosilicates with pores of be obtained by calcination of the ammonium-exchanged molecular dimensions.