GROMACS: Fast, Flexible, and Free
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mercury 2.4 User Guide and Tutorials 2011 CSDS Release
Mercury 2.4 User Guide and Tutorials 2011 CSDS Release Copyright © 2010 The Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The Cambridge Structural Database System (CSD System) comprising all or some of the following: ConQuest, Quest, PreQuest, Mercury, (Mercury CSD and Materials module of Mercury), VISTA, Mogul, IsoStar, SuperStar, web accessible CSD tools and services, WebCSD, CSD Java sketcher, CSD data file, CSD-UNITY, CSD-MDL, CSD-SDfile, CSD data updates, sub files derived from the foregoing data files, documentation and command procedures (each individually a Component) is a database and copyright work belonging to the Cambridge Crystallographic Data Centre (CCDC) and its licensors and all rights are protected. Use of the CSD System is permitted solely in accordance with a valid Licence of Access Agreement and all Components included are proprietary. When a Component is supplied independently of the CSD System its use is subject to the conditions of the separate licence. All persons accessing the CSD System or its Components should make themselves aware of the conditions contained in the Licence of Access Agreement or the relevant licence. In particular: • The CSD System and its Components are licensed subject to a time limit for use by a specified organisation at a specified location. • The CSD System and its Components are to be treated as confidential and may NOT be disclosed or re- distributed in any form, in whole or in part, to any third party. • Software or data derived from or developed using the CSD System may not be distributed without prior written approval of the CCDC. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
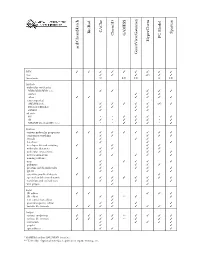
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11 -

Energy Functions and Their Relationship to Molecular Conformation
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5, 2021 Ron Dror 1 Outline • Molecular dynamics (MD): The basic idea • Equations of motion • Key properties of MD simulations • Sample applications • Limitations of MD simulations • Software packages and force fields • Accelerating MD simulations • Monte Carlo simulation 2 Molecular dynamics: The basic idea 3 The idea • Mimic what atoms do in real life, assuming a given potential energy function – The energy function allows us to calculate the force experienced by any atom given the positions of the other atoms – Newton’s laws tell us how those forces will affect the motions of the atoms ) U Energy( Position Position 4 Basic algorithm • Divide time into discrete time steps, no more than a few femtoseconds (10–15 s) each • At each time step: – Compute the forces acting on each atom, using a molecular mechanics force field – Move the atoms a little bit: update position and velocity of each atom using Newton’s laws of motion 5 Molecular dynamics movie Equations of motion 7 Specifying atom positions • For a system with N x1 atoms, we can specify y1 the position of all z1 atoms by a single x2 vector x of length 3N y2 x = – This vector contains z2 the x, y, and z ⋮ coordinates of every xN atom yN zN Specifying forces ∂U • A single vector F specifies F1,x ∂x1 the force acting on every F ∂U atom in the system 1,y ∂y1 • For a system with N F ∂U atoms, F is a vector of 1,z ∂z1 length 3N ∂U F2,x – This vector lists the force ∂x2 ∂U on each atom in the x-, y-, F2,y and z- directions F -

Molecular Docking : Current Advances and Challenges
ARTÍCULO DE REVISIÓN © 2018 Universidad Nacional Autónoma de México, Facultad de Estudios Superiores Zaragoza. This is an Open Access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). TIP Revista Especializada en Ciencias Químico-Biológicas, 21(Supl. 1): 65-87, 2018. DOI: 10.22201/fesz.23958723e.2018.0.143 MOLECULAR DOCKING: CURRENT ADVANCES AND CHALLENGES Fernando D. Prieto-Martínez,1a Marcelino Arciniega2 and José L. Medina-Franco1b 1Facultad de Química, Departamento de Farmacia, Universidad Nacional Autónoma de México, Avenida Universidad # 3000, Delegación Coyoacán, Ciudad de México 04510, México 2Instituto de Fisiología Celular, Departamento de Bioquímica y Biología Estructural, Universidad Nacional Autónoma de México E-mails: [email protected], [email protected],[email protected] ABSTRACT Automated molecular docking aims at predicting the possible interactions between two molecules. This method has proven useful in medicinal chemistry and drug discovery providing atomistic insights into molecular recognition. Over the last 20 years methods for molecular docking have been improved, yielding accurate results on pose prediction. Nonetheless, several aspects of molecular docking need revision due to changes in the paradigm of drug discovery. In the present article, we review the principles, techniques, and algorithms for docking with emphasis on protein-ligand docking for drug discovery. We also discuss current approaches to address major challenges of docking. Key Words: chemoinformatics, computer-aided drug design, drug discovery, structure-activity relationships. Acoplamiento Molecular: Avances Recientes y Retos RESUMEN El acoplamiento molecular automatizado tiene como objetivo proponer un modelo de unión entre dos moléculas. Este método ha sido útil en química farmacéutica y en el descubrimiento de nuevos fármacos por medio del entendimiento de las fuerzas de interacción involucradas en el reconocimiento molecular. -

Downloaded From: Usage Rights: Creative Commons: Attribution-Noncommercial-No Deriva- Tive Works 4.0
Simbanegavi, Nyevero Abigail (2014) An integrated computational and ex- perimental approach to designing novel nanodevices. Doctoral thesis (PhD), Manchester Metropolitan University. Downloaded from: https://e-space.mmu.ac.uk/620241/ Usage rights: Creative Commons: Attribution-Noncommercial-No Deriva- tive Works 4.0 Please cite the published version https://e-space.mmu.ac.uk AN INTEGRATED COMPUTATIONAL AND EXPERIMENTAL APPROACH TO DESIGNING NOVEL NANODEVICES N. A. SIMBANEGAVI PhD 2014 AN INTEGRATED COMPUTATIONAL AND EXPERIMENTAL APPROACH TO DESIGNING NOVEL NANODEVICES Nyevero Abigail Simbanegavi A thesis submitted in partial fulfilment of the requirements of Manchester Metropolitan University for the degree of Doctor of Philosophy 2014 School of Science and the Environment Division of Chemistry and Environmental Science Manchester Metropolitan University Abstract Small molecules that can organise themselves through reversible bonds to form new molecular structures have great potential to form self-assembling systems. In this study, C60 has been functionalized with components capable of self-assembling via hydrogen bonds in order to form supramolecular structures. Functionalizing the cage not only enhances the solubility of C60 but also alters its electronic properties. In order to analyse the changes in electronic properties of C60 and those of the new derivatives and self-assembled structures, an integrated computational and experimental approach has been used. DNA bases were among the components chosen for functionalizing C60 since these structures are the best example for self-assembly in nature. Experimental routes for novel C60 derivatives functionalised with thymine, adenine, cytosine and guanine have been explored using the Prato reaction. A number of challenges in synthesizing the aldehyde analogues of the DNA bases have been identified, and a number of different solutions have been tested. -

Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs
marine drugs Review Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs Guilin Chen 1,2,3, Armel Jackson Seukep 1,2,3,4 and Mingquan Guo 1,2,3,* 1 Key Laboratory of Plant Germplasm Enhancement & Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan 430074, China; [email protected] (G.C.); [email protected] (A.J.S.) 2 Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan 430074, China 3 Innovation Academy for Drug Discovery and Development, Chinese Academy of Sciences, Shanghai 201203, China 4 Department of Biomedical Sciences, Faculty of Health Sciences, University of Buea, P.O. Box 63 Buea, Cameroon * Correspondence: [email protected]; Tel.: +86-27-8770-0850 Received: 16 September 2020; Accepted: 28 October 2020; Published: 30 October 2020 Abstract: Marine drugs have long been used and exhibit unique advantages in clinical practices. Among the marine drugs that have been approved by the Food and Drug Administration (FDA), the protein–ligand interactions, such as cytarabine–DNA polymerase, vidarabine–adenylyl cyclase, and eribulin–tubulin complexes, are the important mechanisms of action for their efficacy. However, the complex and multi-targeted components in marine medicinal resources, their bio-active chemical basis, and mechanisms of action have posed huge challenges in the discovery and development of marine drugs so far, which need to be systematically investigated in-depth. Molecular docking could effectively predict the binding mode and binding energy of the protein–ligand complexes and has become a major method of computer-aided drug design (CADD), hence this powerful tool has been widely used in many aspects of the research on marine drugs. -

Visualization and Analysis of Petascale Molecular Simulations with VMD John E
S4410: Visualization and Analysis of Petascale Molecular Simulations with VMD John E. Stone Theoretical and Computational Biophysics Group Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign http://www.ks.uiuc.edu/Research/gpu/ S4410, GPU Technology Conference 15:30-16:20, Room LL21E, San Jose Convention Center, San Jose, CA, Tuesday March 25, 2014 Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics Beckman Institute, University of Illinois at Urbana-Champaign - www.ks.uiuc.edu VMD – “Visual Molecular Dynamics” • Visualization and analysis of: – molecular dynamics simulations – particle systems and whole cells – cryoEM densities, volumetric data – quantum chemistry calculations – sequence information • User extensible w/ scripting and plugins Whole Cell Simulation MD Simulations • http://www.ks.uiuc.edu/Research/vmd/ CryoEM, Cellular Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics SequenceBeckman Data Institute, University of Illinois at QuantumUrbana-Champaign Chemistry - www.ks.uiuc.edu Tomography Goal: A Computational Microscope Study the molecular machines in living cells Ribosome: target for antibiotics Poliovirus Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics Beckman Institute, University of Illinois at Urbana-Champaign - www.ks.uiuc.edu VMD Interoperability Serves Many Communities • VMD 1.9.1 user statistics: – 74,933 unique registered users from all over the world • Uniquely interoperable