Visualization and Analysis of Petascale Molecular Simulations with VMD John E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
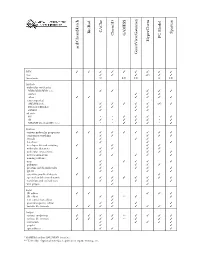
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11 -

Porting the DFT Code CASTEP to Gpgpus
Porting the DFT code CASTEP to GPGPUs Toni Collis [email protected] EPCC, University of Edinburgh CASTEP and GPGPUs Outline • Why are we interested in CASTEP and Density Functional Theory codes. • Brief introduction to CASTEP underlying computational problems. • The OpenACC implementation http://www.nu-fuse.com CASTEP: a DFT code • CASTEP is a commercial and academic software package • Capable of Density Functional Theory (DFT) and plane wave basis set calculations. • Calculates the structure and motions of materials by the use of electronic structure (atom positions are dictated by their electrons). • Modern CASTEP is a re-write of the original serial code, developed by Universities of York, Durham, St. Andrews, Cambridge and Rutherford Labs http://www.nu-fuse.com CASTEP: a DFT code • DFT/ab initio software packages are one of the largest users of HECToR (UK national supercomputing service, based at University of Edinburgh). • Codes such as CASTEP, VASP and CP2K. All involve solving a Hamiltonian to explain the electronic structure. • DFT codes are becoming more complex and with more functionality. http://www.nu-fuse.com HECToR • UK National HPC Service • Currently 30- cabinet Cray XE6 system – 90,112 cores • Each node has – 2×16-core AMD Opterons (2.3GHz Interlagos) – 32 GB memory • Peak of over 800 TF and 90 TB of memory http://www.nu-fuse.com HECToR usage statistics Phase 3 statistics (Nov 2011 - Apr 2013) Ab initio codes (VASP, CP2K, CASTEP, ONETEP, NWChem, Quantum Espresso, GAMESS-US, SIESTA, GAMESS-UK, MOLPRO) GS2NEMO ChemShell 2%2% SENGA2% 3% UM Others 4% 34% MITgcm 4% CASTEP 4% GROMACS 6% DL_POLY CP2K VASP 5% 8% 19% http://www.nu-fuse.com HECToR usage statistics Phase 3 statistics (Nov 2011 - Apr 2013) 35% of the Chemistry software on HECToR is using DFT methods. -
Massive-Parallel Implementation of the Resolution-Of-Identity Coupled
Article Cite This: J. Chem. Theory Comput. 2019, 15, 4721−4734 pubs.acs.org/JCTC Massive-Parallel Implementation of the Resolution-of-Identity Coupled-Cluster Approaches in the Numeric Atom-Centered Orbital Framework for Molecular Systems † § † † ‡ § § Tonghao Shen, , Zhenyu Zhu, Igor Ying Zhang,*, , , and Matthias Scheffler † Department of Chemistry, Fudan University, Shanghai 200433, China ‡ Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, MOE Key Laboratory of Computational Physical Science, Fudan University, Shanghai 200433, China § Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany *S Supporting Information ABSTRACT: We present a massive-parallel implementation of the resolution of identity (RI) coupled-cluster approach that includes single, double, and perturbatively triple excitations, namely, RI-CCSD(T), in the FHI-aims package for molecular systems. A domain-based distributed-memory algorithm in the MPI/OpenMP hybrid framework has been designed to effectively utilize the memory bandwidth and significantly minimize the interconnect communication, particularly for the tensor contraction in the evaluation of the particle−particle ladder term. Our implementation features a rigorous avoidance of the on- the-fly disk storage and excellent strong scaling of up to 10 000 and more cores. Taking a set of molecules with different sizes, we demonstrate that the parallel performance of our CCSD(T) code is competitive with the CC implementations in state-of- the-art high-performance-computing computational chemistry packages. We also demonstrate that the numerical error due to the use of RI approximation in our RI-CCSD(T) method is negligibly small. Together with the correlation-consistent numeric atom-centered orbital (NAO) basis sets, NAO-VCC-nZ, the method is applied to produce accurate theoretical reference data for 22 bio-oriented weak interactions (S22), 11 conformational energies of gaseous cysteine conformers (CYCONF), and 32 Downloaded via FRITZ HABER INST DER MPI on January 8, 2021 at 22:13:06 (UTC). -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title From NWChem to NWChemEx: Evolving with the Computational Chemistry Landscape. Permalink https://escholarship.org/uc/item/4sm897jh Journal Chemical reviews, 121(8) ISSN 0009-2665 Authors Kowalski, Karol Bair, Raymond Bauman, Nicholas P et al. Publication Date 2021-04-01 DOI 10.1021/acs.chemrev.0c00998 Peer reviewed eScholarship.org Powered by the California Digital Library University of California From NWChem to NWChemEx: Evolving with the computational chemistry landscape Karol Kowalski,y Raymond Bair,z Nicholas P. Bauman,y Jeffery S. Boschen,{ Eric J. Bylaska,y Jeff Daily,y Wibe A. de Jong,x Thom Dunning, Jr,y Niranjan Govind,y Robert J. Harrison,k Murat Keçeli,z Kristopher Keipert,? Sriram Krishnamoorthy,y Suraj Kumar,y Erdal Mutlu,y Bruce Palmer,y Ajay Panyala,y Bo Peng,y Ryan M. Richard,{ T. P. Straatsma,# Peter Sushko,y Edward F. Valeev,@ Marat Valiev,y Hubertus J. J. van Dam,4 Jonathan M. Waldrop,{ David B. Williams-Young,x Chao Yang,x Marcin Zalewski,y and Theresa L. Windus*,r yPacific Northwest National Laboratory, Richland, WA 99352 zArgonne National Laboratory, Lemont, IL 60439 {Ames Laboratory, Ames, IA 50011 xLawrence Berkeley National Laboratory, Berkeley, 94720 kInstitute for Advanced Computational Science, Stony Brook University, Stony Brook, NY 11794 ?NVIDIA Inc, previously Argonne National Laboratory, Lemont, IL 60439 #National Center for Computational Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831-6373 @Department of Chemistry, Virginia Tech, Blacksburg, VA 24061 4Brookhaven National Laboratory, Upton, NY 11973 rDepartment of Chemistry, Iowa State University and Ames Laboratory, Ames, IA 50011 E-mail: [email protected] 1 Abstract Since the advent of the first computers, chemists have been at the forefront of using computers to understand and solve complex chemical problems. -

Computational Chemistry
Computational Chemistry as a Teaching Aid Melissa Boule Aaron Harshman Rexford Hoadley An Interactive Qualifying Project Report Submitted to the Faculty of the Worcester Polytechnic Institute In partial fulfillment of the requirements for the Bachelor of Science Degree Approved by: Professor N. Aaron Deskins 1i Abstract Molecular modeling software has transformed the capabilities of researchers. Molecular modeling software has the potential to be a helpful teaching tool, though as of yet its efficacy as a teaching technique has yet to be proven. This project focused on determining the effectiveness of a particular molecular modeling software in high school classrooms. Our team researched what topics students struggled with, surveyed current high school chemistry teachers, chose a modeling software, and then developed a lesson plan around these topics. Lastly, we implemented the lesson in two separate high school classrooms. We concluded that these programs show promise for the future, but with the current limitations of high school technology, these tools may not be as impactful. 2ii Acknowledgements This project would not have been possible without the help of many great educators. Our advisor Professor N. Aaron Deskins provided driving force to ensure the research ran smoothly and efficiently. Mrs. Laurie Hanlan and Professor Drew Brodeur provided insight and guidance early in the development of our topic. Mr. Mark Taylor set-up our WebMO server and all our other technology needs. Lastly, Mrs. Pamela Graves and Mr. Eric Van Inwegen were instrumental by allowing us time in their classrooms to implement our lesson plans. Without the support of this diverse group of people this project would never have been possible. -

1 Advances in Electronic Structure Theory: Gamess a Decade Later Mark S. Gordon and Michael W. Schmidt Department of Chemistry A
ADVANCES IN ELECTRONIC STRUCTURE THEORY: GAMESS A DECADE LATER MARK S. GORDON AND MICHAEL W. SCHMIDT DEPARTMENT OF CHEMISTRY AND AMES LABORATORY, IOWA STATE UNIVERSITY, AMES, IA 50011 Abstract. Recent developments in advanced quantum chemistry and quantum chemistry interfaced with model potentials are discussed, with the primary focus on new implementations in the GAMESS electronic structure suite of programs. Applications to solvent effects and surface science are discussed. 1. Introduction The past decade has seen an extraordinary growth in novel new electronic structure methods and creative implementations of these methods. Concurrently, there have been important advances in “middleware”, software that enables the implementation of efficient electronic structure algorithms. Combined with continuing improvements in computer and interconnect hardware, these advances have extended both the accuracy of computations and the sizes of molecular systems to which such methods may be applied. The great majority of the new computational chemistry algorithms have found their way into one or more broadly distributed electronic structure packages. Since this work focuses on new features of the GAMESS (General Atomic and Molecular Electronic Structure System1) suite of codes, it is important at the outset to recognize the many other packages that offer both similar and complementary features. These include ACES2 CADPAC3, DALTON4, GAMESS-UK5, HYPERCHEM6, JAGUAR7, MOLCAS8 MOLPRO9, NWChem10, PQS11, PSI312, Q-Chem13, SPARTAN14, TURBOMOLE15, and UT-Chem16. Some1,3,4,10 of these packages are distributed at no cost to all, or to academic users, while others are commercial packages, but all are generally available to users without restriction or constraint. Indeed, the developers of these codes frequently collaborate to share features, a practice that clearly benefits all of their users. -

Manual-2018.Pdf
GROMACS Groningen Machine for Chemical Simulations Reference Manual Version 2018 GROMACS Reference Manual Version 2018 Contributions from Emile Apol, Rossen Apostolov, Herman J.C. Berendsen, Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra, Sebastian Fritsch, Gerrit Groenhof, Christoph Junghans, Jochen Hub, Peter Kasson, Carsten Kutzner, Brad Lambeth, Per Larsson, Justin A. Lemkul, Viveca Lindahl, Magnus Lundborg, Erik Marklund, Pieter Meulenhoff, Teemu Murtola, Szilárd Páll, Sander Pronk, Roland Schulz, Michael Shirts, Alfons Sijbers, Peter Tieleman, Christian Wennberg and Maarten Wolf. Mark Abraham, Berk Hess, David van der Spoel, and Erik Lindahl. c 1991–2000: Department of Biophysical Chemistry, University of Groningen. Nijenborgh 4, 9747 AG Groningen, The Netherlands. c 2001–2018: The GROMACS development teams at the Royal Institute of Technology and Uppsala University, Sweden. More information can be found on our website: www.gromacs.org. iv Preface & Disclaimer This manual is not complete and has no pretention to be so due to lack of time of the contributors – our first priority is to improve the software. It is worked on continuously, which in some cases might mean the information is not entirely correct. Comments on form and content are welcome, please send them to one of the mailing lists (see www.gromacs.org), or open an issue at redmine.gromacs.org. Corrections can also be made in the GROMACS git source repository and uploaded to gerrit.gromacs.org. We release an updated version of the manual whenever we release a new version of the software, so in general it is a good idea to use a manual with the same major and minor release number as your GROMACS installation.