Parameterizing a Novel Residue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MODELLER 10.1 Manual
MODELLER A Program for Protein Structure Modeling Release 10.1, r12156 Andrej Saliˇ with help from Ben Webb, M.S. Madhusudhan, Min-Yi Shen, Guangqiang Dong, Marc A. Martı-Renom, Narayanan Eswar, Frank Alber, Maya Topf, Baldomero Oliva, Andr´as Fiser, Roberto S´anchez, Bozidar Yerkovich, Azat Badretdinov, Francisco Melo, John P. Overington, and Eric Feyfant email: modeller-care AT salilab.org URL https://salilab.org/modeller/ 2021/03/12 ii Contents Copyright notice xxi Acknowledgments xxv 1 Introduction 1 1.1 What is Modeller?............................................. 1 1.2 Modeller bibliography....................................... .... 2 1.3 Obtainingandinstallingtheprogram. .................... 3 1.4 Bugreports...................................... ............ 4 1.5 Method for comparative protein structure modeling by Modeller ................... 5 1.6 Using Modeller forcomparativemodeling. ... 8 1.6.1 Preparinginputfiles . ............. 8 1.6.2 Running Modeller ......................................... 9 2 Automated comparative modeling with AutoModel 11 2.1 Simpleusage ..................................... ............ 11 2.2 Moreadvancedusage............................... .............. 12 2.2.1 Including water molecules, HETATM residues, and hydrogenatoms .............. 12 2.2.2 Changing the default optimization and refinement protocol ................... 14 2.2.3 Getting a very fast and approximate model . ................. 14 2.2.4 Building a model from multiple templates . .................. 15 2.2.5 Buildinganallhydrogenmodel -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -
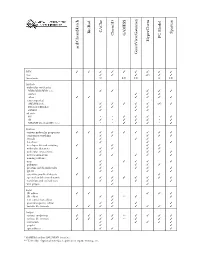
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11 -

Downloaded From: Usage Rights: Creative Commons: Attribution-Noncommercial-No Deriva- Tive Works 4.0
Simbanegavi, Nyevero Abigail (2014) An integrated computational and ex- perimental approach to designing novel nanodevices. Doctoral thesis (PhD), Manchester Metropolitan University. Downloaded from: https://e-space.mmu.ac.uk/620241/ Usage rights: Creative Commons: Attribution-Noncommercial-No Deriva- tive Works 4.0 Please cite the published version https://e-space.mmu.ac.uk AN INTEGRATED COMPUTATIONAL AND EXPERIMENTAL APPROACH TO DESIGNING NOVEL NANODEVICES N. A. SIMBANEGAVI PhD 2014 AN INTEGRATED COMPUTATIONAL AND EXPERIMENTAL APPROACH TO DESIGNING NOVEL NANODEVICES Nyevero Abigail Simbanegavi A thesis submitted in partial fulfilment of the requirements of Manchester Metropolitan University for the degree of Doctor of Philosophy 2014 School of Science and the Environment Division of Chemistry and Environmental Science Manchester Metropolitan University Abstract Small molecules that can organise themselves through reversible bonds to form new molecular structures have great potential to form self-assembling systems. In this study, C60 has been functionalized with components capable of self-assembling via hydrogen bonds in order to form supramolecular structures. Functionalizing the cage not only enhances the solubility of C60 but also alters its electronic properties. In order to analyse the changes in electronic properties of C60 and those of the new derivatives and self-assembled structures, an integrated computational and experimental approach has been used. DNA bases were among the components chosen for functionalizing C60 since these structures are the best example for self-assembly in nature. Experimental routes for novel C60 derivatives functionalised with thymine, adenine, cytosine and guanine have been explored using the Prato reaction. A number of challenges in synthesizing the aldehyde analogues of the DNA bases have been identified, and a number of different solutions have been tested. -

Computational Chemistry (F14CCH)
Computational Chemistry (F14CCH) Connecting to Remote Machines Using CCP1GUI CONDOR is a queuing system that can be accessed from the Linux Left Mouse Button: Turn machines of the theoretical research groups and distributes the Hold right + up or down: Zoom submitted jobs to free compute nodes. Since the machines are protected Hold wheel + move: Move by firewalls you have to connect to a specific node, Porthos, on which you were given an account. Click Atom => Select Fragment => Add Assign all elements, also the hydrogens, before creating the GAMESS Use PuTTY to connect to remote machines, porthos in this case. Open input file (click on “All X => H” in the tool panel). If you cannot see one of putty.exe, enter the hostname as porthos.chem.nott.ac.uk. The login the windows of CCP1GUI, it might be hiding outside the desktop area. is your university username and the default password is Right click on the taskbar icon => Move => hold cursor keys to the left ChangeMeSoon. When you first log in on CONDOR you must change until the window appears. your password using kpasswd! It must be strong, including mixed case letters and numbers. Creating a GAMESS Input File / Running a Local Job Compute => GAMESS-UK Copying Files from/to Porthos To exchange files with porthos in order to submit files to CONDOR for Molecule: Options => Title calculation or copy results back, you first have to copy them from the Task => select requested task Windows machine to porthos via the SSHClient. It cannot be installed in Theory: Select SCF Method (and post SCF Method, if required) the CAL but can be accessed at NAL => Accessing the Internet => Optimisation: Runtype => Opt. -

Molecular Simulation and Structure Prediction Using CHARMM and the MMTSB Tool Set Introduction
Molecular simulation and structure prediction using CHARMM and the MMTSB Tool Set Introduction Charles L. Brooks III MMTSB/CTBP 2006 Summer Workshop MMTSB/CTBP Summer Workshop © Charles L. Brooks III, 2006. CTBP and MMTSB: Who are we? • The CTBP is the Center for Theoretical Biological Physics – Funded by the NSF as a Physics Frontiers Center – Partnership between UCSD, Scripps and Salk, lead by UCSD • The CTBP encompasses a broad spectrum of research and training activities at the forefront of the biology- physics interface. – Principal scientists include • José Onuchic, Herbie Levine, Henry Abarbanel, Charles Brooks, David Case, Mike Holst, Terry Hwa, David Kleinfeld, Andy McCammon, Wouter Rappel, Terry Sejnowski, Wei Wang, Peter Wolynes MMTSB/CTBP Summer Workshop © Charles L. Brooks III, 2006. CTBP and MMTSB: Who are we? MMTSB/CTBP Summer Workshop http://ctbp.ucsd.edu © Charles L. Brooks III, 2006. CTBP and MMTSB: Who are we? • The MMTSB is the Center for Multi-scale Modeling Tools for Structural Biology – Funded by the NIH as a National Research Resource Center – Partnership between Scripps and Georgia Tech, lead by Scripps • The MMTSB aims to develop new tools and theoretical models to aid molecular and structural biologists in interpreting their biological data. – Principal scientists include • Charles Brooks, David Case, Steve Harvey, Jack Johnson, Vijay Reddy, Jeff Skolnick MMTSB/CTBP Summer Workshop © Charles L. Brooks III, 2006. CTBP and MMTSB: Who are we? http://mmtsb.scripps.edu MMTSB/CTBP Summer Workshop © Charles L. Brooks III, 2006. CTBP and MMTSB: Who are we? • Activities – Fundamental research across a broad spectrum – Software and methods development and distribution • MMTSB distributes multiple software packages as well as hosts a variety of web services and databases – Training and research workshops and educational outreach • Both centers have extensive workshop programs – Visitors • Both centers host visitors and collaborators for short and longer term (sabbatical) visits MMTSB/CTBP Summer Workshop © Charles L. -

Visualization and Analysis of Petascale Molecular Simulations with VMD John E
S4410: Visualization and Analysis of Petascale Molecular Simulations with VMD John E. Stone Theoretical and Computational Biophysics Group Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign http://www.ks.uiuc.edu/Research/gpu/ S4410, GPU Technology Conference 15:30-16:20, Room LL21E, San Jose Convention Center, San Jose, CA, Tuesday March 25, 2014 Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics Beckman Institute, University of Illinois at Urbana-Champaign - www.ks.uiuc.edu VMD – “Visual Molecular Dynamics” • Visualization and analysis of: – molecular dynamics simulations – particle systems and whole cells – cryoEM densities, volumetric data – quantum chemistry calculations – sequence information • User extensible w/ scripting and plugins Whole Cell Simulation MD Simulations • http://www.ks.uiuc.edu/Research/vmd/ CryoEM, Cellular Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics SequenceBeckman Data Institute, University of Illinois at QuantumUrbana-Champaign Chemistry - www.ks.uiuc.edu Tomography Goal: A Computational Microscope Study the molecular machines in living cells Ribosome: target for antibiotics Poliovirus Biomedical Technology Research Center for Macromolecular Modeling and Bioinformatics Beckman Institute, University of Illinois at Urbana-Champaign - www.ks.uiuc.edu VMD Interoperability Serves Many Communities • VMD 1.9.1 user statistics: – 74,933 unique registered users from all over the world • Uniquely interoperable