Chemdraw V19.0 User Guide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pre-Activity Quiz Answer Key (Pdf)
Name: ____________________________________________ Date: ___________________ Class: ________________________ Pre-Activity Quiz Answer Key 1. Consider a molecule of carbon monoxide (C O): A. Do you think the electrons in the triple bond pull closer to the C atom or the O atom, or are they equally shared? Use the concept of electronegativity to explain your response. Answer: They are closer to the O atom. Using the electronegativity scale from a textbook, the result is a polar covalent bond shifted towards the O atom. B. Is the bond polar or non-polar? Polar 2. In today’s engineering challenge, you will sketch out Lewis dot diagrams for various molecules and polyatomic ions. Then you will construct each molecule using a molecular model kit. The kits contain three different representations: colored balls, short sticks and long flexible springs. A. Each colored ball corresponds to a different atom. How can you determine which color to use for each atom? Use the reference sheet that comes with the kit. B. For what bond type do you think the short sticks are used? Covalent bonds C. If you were to build a triple bond, what would you use to represent a triple bond and how many would you use? Use springs, three springs 3. You will become familiar with different geometries of simple molecules. A. Name the theory used to predict molecular shapes of these molecules? VSEPR theory Molecular Models and 3-D Printing Activity—Pre-Activity Quiz Answer Key 1 Name: ____________________________________________ Date: ___________________ Class: ________________________ B. What if a molecule contains a central atom bonded to two identical outer atoms with the central atom surrounded by a lone pair of electrons? Name the geometry of this molecule. -

Nextfit ® Ix Car Seat
Convertible Car Seat User Guide For future use, STORE USER GUIDE in compartment at rear of base. ©2016 Artsana USA, Inc. IS0148.3E www.chiccousa.com If you have any problems with your Chicco Child Restraint, or any questions regarding installation or use, please call: Chicco Customer Service 1-877-424-4226 Please have Model and Serial Number available when you call. These are located on a label on the bottom of the Child Restraint. For future reference, fill in the information below. The information can be found on the label on the bottom of the Child Restraint. Model Number: Serial Number: Manufactured In: TABLE OF CONTENTS Registration and Recall 2 REAR-FACING INSTALLATION Child Guidelines 4 Rear-Facing Setup 38 Safe Use Checklist 6 Install Using LATCH 42 Important Warnings 8 Install Using LAP-SHOULDER BELT 48 Best Practices 14 Install Using LAP BELT ONLY 54 Need Help? 15 FORWARD-FACING INSTALLATION CHILD RESTRAINT OVERVIEW Forward-Facing Setup 60 Child Restraint Components 16 Install Using LATCH 64 LATCH and Tether Components 18 Install Using LAP-SHOULDER BELT 70 LATCH and Tether Storage 20 Install Using LAP BELT ONLY 76 Selecting Rear/Forward Facing Position 22 Adjusting Crotch Strap 26 SECURING YOUR CHILD Newborn Insert 28 Securing Child with Harness 80 Secure Child Checklist 92 VEHICLE INFORMATION Vehicle Seating Positions 30 ADDITIONAL INFORMATION Vehicle Seat Belts 32 Installation on an Aircraft 94 What is LATCH? 34 Cup Holder 96 What is a Tether? 36 Cleaning and Maintenance 98 REGISTRATION AND RECALL Please complete the Registration Card that came with your Child Restraint and mail it promptly. -
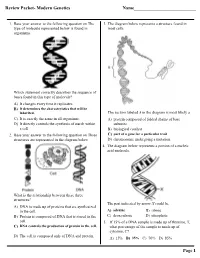
Review Packet- Modern Genetics Name___Page 1
Review Packet- Modern Genetics Name___________________________ 1. Base your answer to the following question on The 3. The diagram below represents a structure found in type of molecule represented below is found in most cells. organisms. Which statement correctly describes the sequence of bases found in this type of molecule? A) It changes every time it replicates. B) It determines the characteristics that will be inherited. The section labeled A in the diagram is most likely a C) It is exactly the same in all organisms. A) protein composed of folded chains of base D) It directly controls the synthesis of starch within subunits a cell. B) biological catalyst 2. Base your answer to the following question on Three C) part of a gene for a particular trait structures are represented in the diagram below. D) chromosome undergoing a mutation 4. The diagram below represents a portion of a nucleic acid molecule. What is the relationship between these three structures? The part indicated by arrow X could be A) DNA is made up of proteins that are synthesized in the cell. A) adenine B) ribose B) Protein is composed of DNA that is stored in the C) deoxyribose D) phosphate cell. 5. If 15% of a DNA sample is made up of thymine, T, C) DNA controls the production of protein in the cell. what percentage of the sample is made up of cytosine, C? D) The cell is composed only of DNA and protein. A) 15% B) 35% C) 70% D) 85% Page 1 6. Base your answer to the following question on Which 10.The diagram below represents genetic material. -

Significance and Implications of Vitamin B-12 Reaction Shema- ETH ZURICH VARIANT: Mechanisms and Insights
Taylor University Pillars at Taylor University Student Scholarship: Chemistry Chemistry and Biochemistry Fall 2019 Significance and Implications of Vitamin B-12 Reaction Shema- ETH ZURICH VARIANT: Mechanisms and Insights David Joshua Ferguson Follow this and additional works at: https://pillars.taylor.edu/chemistry-student Part of the Analytical Chemistry Commons, Inorganic Chemistry Commons, Organic Chemistry Commons, Other Chemistry Commons, and the Physical Chemistry Commons CHEMISTRY THESIS SIGNIFICANCE AND IMPLICATIONS OF VITAMIN B-12 REACTION SCHEMA- ETH ZURICH VARIANT: MECHANISMS AND INSIGHTS DAVID JOSHUA FERGUSON 2019 2 Table of Contents: Chapter 1 6 Chapter 2 17 Chapter 3 40 Chapter 4 59 Chapter 5 82 Chapter 6 118 Chapter 7 122 Appendix References 3 Chapter 1 A. INTRODUCTION. Vitamin B-12 otherwise known as cyanocobalamin is a compound with synthetic elegance. Considering how it is composed of an aromatic macrocyclic corrin there are key features of this molecule that are observed either in its synthesis of in the biochemical reactions it plays a role in whether they be isomerization reactions or transfer reactions. In this paper the focus for the discussion will be on the history, chemical significance and total synthesis of vitamin B12. Even more so the paper will be concentrated one of the two variants of the vitamin B-12 synthesis, namely the ETH Zurich variant spearheaded by Albert Eschenmoser.Examining the structure as a whole it is observed that a large portion of the vitamin B12 is a corrin structure with a cobalt ion in the center of the macrocyclic part, and that same cobalt ion has cyanide ligands. -

Rare-Earth Metal Methylidene Complexes with Ln3 (Μ3-CH2)(Μ3
Dalton Transactions View Article Online PAPER View Journal | View Issue Rare-earth metal methylidene complexes with Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 core structure† Cite this: Dalton Trans., 2015, 44, 18101 Dorothea Schädle,a Melanie Meermann-Zimmermann,b Cäcilia Maichle-Mössmer,a Christoph Schädle,a Karl W. Törnroosc and Reiner Anwander*a Trinuclear rare-earth metal methylidene complexes with a Ln3(µ3-CH2)(µ3-Me)(µ2-Me)3 structural motif were synthesized by applying three protocols. Polymeric [LuMe3]n (1-Lu) reacts with the sterically demand- ing amine H[NSiMe3(Ar)] (Ar = C6H3iPr2-2,6) in tetrahydrofuran via methane elimination to afford isolable monomeric [NSiMe3(Ar)]LuMe2(thf)2 (4-Lu). The formation of trinuclear rare-earth metal tetramethyl methylidene complexes [NSiMe3(Ar)]3Ln3(µ3-CH2)(µ3-Me)(µ2-Me)3(thf)3 (7-Ln; Ln = Y, Ho, Lu) via reaction of [LnMe3]n (1-Ln; Ln = Y, Ho, Lu) with H[NSiMe3(Ar)] is proposed to occur via an “intermediate” species of the type [NSiMe3(Ar)]LnMe2(thf)x and subsequent C−H bond activation. Applying Lappert’s concept of Lewis base-induced methylaluminate cleavage, compounds [NSiMe3(Ar)]Ln(AlMe4)2 (5-Ln; Ln = Y, La, Nd, Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Ho) were converted into methylidene complexes 7-Ln (Ln = Y, Nd, Ho) in the presence of tetrahydrofuran. Similarly, tetramethylgallate complex [NSiMe3(Ar)]Y(GaMe4)2 (6-Y) could be employed as a synthesis pre- cursor for 7-Y. The molecular composition of complexes 4-Ln, 5-Ln, 6-Y and 7-Ln was confirmed by elemental analyses, FTIR spectroscopy, 1H and 13C NMR spectroscopy (except for holmium derivatives) Received 30th July 2015, and single-crystal X-ray diffraction. -

Solving the Schrödinger Equation for Helium Atom and Its Isoelectronic
THE JOURNAL OF CHEMICAL PHYSICS 127, 224104 ͑2007͒ Solving the Schrödinger equation for helium atom and its isoelectronic ions with the free iterative complement interaction „ICI… method ͒ Hiroyuki Nakashima and Hiroshi Nakatsujia Quantum Chemistry Research Institute, Kyodai Katsura Venture Plaza 106, Goryo Oohara 1-36, Nishikyo-ku, Kyoto 615-8245, Japan and Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan ͑Received 31 July 2007; accepted 2 October 2007; published online 11 December 2007͒ The Schrödinger equation was solved very accurately for helium atom and its isoelectronic ions ͑Z=1–10͒ with the free iterative complement interaction ͑ICI͒ method followed by the variational principle. We obtained highly accurate wave functions and energies of helium atom and its isoelectronic ions. For helium, the calculated energy was −2.903 724 377 034 119 598 311 159 245 194 404 446 696 905 37 a.u., correct over 40 digit accuracy, and for H−,itwas−0.527 751 016 544 377 196 590 814 566 747 511 383 045 02 a.u. These results prove numerically that with the free ICI method, we can calculate the solutions of the Schrödinger equation as accurately as one desires. We examined several types of scaling function g and initial function 0 of the free ICI method. The performance was good when logarithm functions were used in the initial function because the logarithm function is physically essential for three-particle collision area. The best performance was obtained when we introduce a new logarithm function containing not only r1 and r2 but also r12 in the same logarithm function. -

Many-Electron System I – Helium Atom and Pauli Exclusion Principle
Lecture 17 Many-electron System I { Helium Atom and Pauli Exclusion Principle Study Goal of This Lecture • Helium atom - many electron wavefunctions • Helium atom - variational ground state (screening) • Eigenstates of a two-spin system 17.1 Helium Atom Helium atom includes two electrons moving around a fixed nucleus with charge Z = 2, we can write down the Hamiltonian as: 2 2 2 2 ^ ~ 2 2 1 Ze Ze e H = − (r1 + r2) − ( + − ) 2me 4π0 r1 r2 r12 2 2 2 2 2 ~ 2 1 Ze ~ 2 1 Ze e = − r1 − − − r2 − + 2me 4π0 r1 2me 4π0 r2 4π0r12 (17.1) | {z } | {z } | {z } H^1 H^2 H^12 = H^1 + H^2 + H^12: 1 If H^12 = 0(or neglected) then the problem is exactly solved. Recall that for a total system composes of independent sub-systems H^T = H^1 + H^2 + ··· ; (17.2) and we can firstly solve all ^ n n n Hnφm = Emφm; (17.3) then the solution of H^T is the product states Y n = φ : (17.4) n If consider as many-electrons, i.e. H^n for the n-th electron. Then the product solu- tion is a natural "independent electron" solution. Even when the electron-electron interactions are non-zero, we will see the independent electorn approximation is a good starting point. Let's consider the Helium atom, we know: H^1φ1 = E1φ1; (17.5) H^2φ2 = E2φ2; and total E = E1 + E2. φ1; φ2 are Helium hydrogen-like atomic orbitals. We know the two ground states (neglect spin for a moment.) H^1φ1s(1) = E1sφ1s(1); (17.6) H^2φ1s(2) = E1sφ1s(2); number 1 and 2 denotes electorn 1 and electron 2 respectively. -

Identification of a Novel Substrate-Derived Spermine
ЭКСПЕРИМЕНТАЛЬНЫЕ СТАТЬИ UDK 577.152.1 Identification of a Novel Substrate- Derived Spermine Oxidase Inhibitor T. T. Dunston1, M. A. Khomutov2, S. B. Gabelli1,3,4, T. M. Stewart1, J. R. Foley1, S. N. Kochetkov2, A. R. Khomutov2*, R. A. Casero Jr.1* 1Sidney Kimmel Comprehensive Cancer Center, The Johns Hopkins University School of Medicine, Baltimore, MD 21287 USA 2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, 119991 Russia 3Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 4Department of Oncology, The Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA *E-mail: [email protected], [email protected] Received May 08, 2020; in final form, July 07, 2020 DOI: 10.32607/actanaturae.10992 ABSTRACT Homeostasis of the biogenic polyamines spermine (Spm) and spermidine (Spd), present in μM-mM concentrations in all eukaryotic cells, is precisely regulated by coordinated activities of the enzymes of poly- amine synthesis, degradation, and transport, in order to sustain normal cell growth and viability. Spermine oxidase (SMOX) is the key and most recently discovered enzyme of polyamine metabolism that plays an es- sential role in regulating polyamine homeostasis by catalyzing the back-conversion of Spm to Spd. The deve- lopment of many types of epithelial cancer is associated with inflammation, and disease-related inflammatory stimuli induce SMOX. MDL72527 is widely used in vitro and in vivo as an irreversible inhibitor of SMOX, but it is also potent towards N1-acetylpolyamine oxidase. Although SMOX has high substrate specificity, Spm analogues have not been systematically studied as enzyme inhibitors. -

Designing Universal Chemical Markup (UCM) Through the Reusable Methodology Based on Analyzing Existing Related Formats
Designing Universal Chemical Markup (UCM) through the reusable methodology based on analyzing existing related formats Background: In order to design concepts for a new general-purpose chemical format we analyzed the strengths and weaknesses of current formats for common chemical data. While the new format is discussed more in the next article, here we describe our software s t tools and two stage analysis procedure that supplied the necessary information for the n i r development. The chemical formats analyzed in both stages were: CDX, CDXML, CML, P CTfile and XDfile. In addition the following formats were included in the first stage only: e r P CIF, InChI, NCBI ASN.1, NCBI XML, PDB, PDBx/mmCIF, PDBML, SMILES, SLN and Mol2. Results: A two stage analysis process devised for both XML (Extensible Markup Language) and non-XML formats enabled us to verify if and how potential advantages of XML are utilized in the widely used general-purpose chemical formats. In the first stage we accumulated information about analyzed formats and selected the formats with the most general-purpose chemical functionality for the second stage. During the second stage our set of software quality requirements was used to assess the benefits and issues of selected formats. Additionally, the detailed analysis of XML formats structure in the second stage helped us to identify concepts in those formats. Using these concepts we came up with the concise structure for a new chemical format, which is designed to provide precise built-in validation capabilities and aims to avoid the potential issues of analyzed formats. -

Cisco Unified IP Phone Services Application Development Notes Supporting XML Applications
Cisco Unified IP Phone Services Application Development Notes Supporting XML Applications Release 8.5(1) Americas Headquarters Cisco Systems, Inc. 170 West Tasman Drive San Jose, CA 95134-1706 USA http://www.cisco.com Tel: 408 526-4000 800 553-NETS (6387) Fax: 408 527-0883 Text Part Number: OL-22505-01 THE SPECIFICATIONS AND INFORMATION REGARDING THE PRODUCTS IN THIS MANUAL ARE SUBJECT TO CHANGE WITHOUT NOTICE. ALL STATEMENTS, INFORMATION, AND RECOMMENDATIONS IN THIS MANUAL ARE BELIEVED TO BE ACCURATE BUT ARE PRESENTED WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED. USERS MUST TAKE FULL RESPONSIBILITY FOR THEIR APPLICATION OF ANY PRODUCTS. THE SOFTWARE LICENSE AND LIMITED WARRANTY FOR THE ACCOMPANYING PRODUCT ARE SET FORTH IN THE INFORMATION PACKET THAT SHIPPED WITH THE PRODUCT AND ARE INCORPORATED HEREIN BY THIS REFERENCE. IF YOU ARE UNABLE TO LOCATE THE SOFTWARE LICENSE OR LIMITED WARRANTY, CONTACT YOUR CISCO REPRESENTATIVE FOR A COPY. The Cisco implementation of TCP header compression is an adaptation of a program developed by the University of California, Berkeley (UCB) as part of UCB’s public domain version of the UNIX operating system. All rights reserved. Copyright © 1981, Regents of the University of California. NOTWITHSTANDING ANY OTHER WARRANTY HEREIN, ALL DOCUMENT FILES AND SOFTWARE OF THESE SUPPLIERS ARE PROVIDED “AS IS” WITH ALL FAULTS. CISCO AND THE ABOVE-NAMED SUPPLIERS DISCLAIM ALL WARRANTIES, EXPRESSED OR IMPLIED, INCLUDING, WITHOUT LIMITATION, THOSE OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT OR ARISING FROM A COURSE OF DEALING, USAGE, OR TRADE PRACTICE. IN NO EVENT SHALL CISCO OR ITS SUPPLIERS BE LIABLE FOR ANY INDIRECT, SPECIAL, CONSEQUENTIAL, OR INCIDENTAL DAMAGES, INCLUDING, WITHOUT LIMITATION, LOST PROFITS OR LOSS OR DAMAGE TO DATA ARISING OUT OF THE USE OR INABILITY TO USE THIS MANUAL, EVEN IF CISCO OR ITS SUPPLIERS HAVE BEEN ADVISED OF THE POSSIBILITY OF SUCH DAMAGES. -

Notes on OLEX2
Notes on OLEX2 Updated on 12 January 2018,at 09:05. Olex2 v1.2-dev © OlexSys Ltd. 2004 – 2016 Compilation Info: 2017.07.20 svn.r3457 MSC:150030729 on WIN64, Python: 2.7.5, wxWidgets: 3.1.0 for OlexSys Ilia A. Guzei 2124 Chemistry Department, University of Wisconsin-Madison, 1101 University Ave, Madison, WI 53706 USA. This is work in progress. You are encouraged to e-mail me ([email protected]) your comments, corrections, and suggestions. Many thanks to Nattamai Bhuvanesh, Brian Dolinar, Oleg Dolomanov, Dean Johnston, Horst Puschmann, Amy Sarjeant, Charlotte Stern, for proof- reading, suggestions, and comments. I have also borrowed from Martin Lutz, Len Barbour, Richard Staples and Tony Linden. OLEX2 Manual Table of Content Table of Content ........................................................................................................................... 2 How to install OLEX2 under Windows .......................................................................................... 3 How to install OLEX2 on a Mac .................................................................................................... 6 Installing and using PLATON on a Mac ........................................................................................ 8 How to get OLEX2 to use PLATON ............................................................................................ 11 About program OLEX2 ................................................................................................................ 11 Keyboard shortcuts ..................................................................................................................... -

Linking Nwchem and Avogadro with the Syntax and Semantics of Chemical Markup Language Wibe a De Jong1*†, Andrew M Walker2† and Marcus D Hanwell3†
de Jong et al. Journal of Cheminformatics 2013, 5:25 http://www.jcheminf.com/content/5/1/25 RESEARCH ARTICLE Open Access From data to analysis: linking NWChem and Avogadro with the syntax and semantics of Chemical Markup Language Wibe A de Jong1*†, Andrew M Walker2† and Marcus D Hanwell3† Abstract Background: Multidisciplinary integrated research requires the ability to couple the diverse sets of data obtained from a range of complex experiments and computer simulations. Integrating data requires semantically rich information. In this paper an end-to-end use of semantically rich data in computational chemistry is demonstrated utilizing the Chemical Markup Language (CML) framework. Semantically rich data is generated by the NWChem computational chemistry software with the FoX library and utilized by the Avogadro molecular editor for analysis and visualization. Results: The NWChem computational chemistry software has been modified and coupled to the FoX library to write CML compliant XML data files. The FoX library was expanded to represent the lexical input files and molecular orbitals used by the computational chemistry software. Draft dictionary entries and a format for molecular orbitals within CML CompChem were developed. The Avogadro application was extended to read in CML data, and display molecular geometry and electronic structure in the GUI allowing for an end-to-end solution where Avogadro can create input structures, generate input files, NWChem can run the calculation and Avogadro can then read in and analyse the CML output produced. The developments outlined in this paper will be made available in future releases of NWChem, FoX, and Avogadro. Conclusions: The production of CML compliant XML files for computational chemistry software such as NWChem can be accomplished relatively easily using the FoX library.