Enzyme Purificationt GEORGE S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
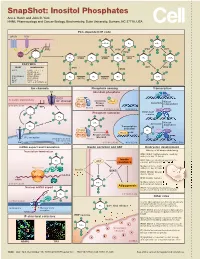
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt
International Journal of Molecular Sciences Article Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt 1,2, 1, 2 2 Stephanie Schwalm y, Martin Erhardt y, Isolde Römer , Josef Pfeilschifter , Uwe Zangemeister-Wittke 1,* and Andrea Huwiler 1,* 1 Institute of Pharmacology, University of Bern, Inselspital, INO-F, CH-3010 Bern, Switzerland; [email protected] (S.S.); [email protected] (M.E.) 2 Institute of General Pharmacology and Toxicology, University Hospital Frankfurt am Main, Goethe-University, Theodor-Stern Kai 7, D-60590 Frankfurt am Main, Germany; [email protected] (I.R.); [email protected] (J.P.) * Correspondence: [email protected] (U.Z.-W.); [email protected] (A.H.); Tel.: +41-31-6323214 (A.H.) These authors contributed equally to this work. y Received: 29 November 2019; Accepted: 12 February 2020; Published: 19 February 2020 Abstract: Ceramide kinase (CerK) is a lipid kinase that converts the proapoptotic ceramide to ceramide 1-phosphate, which has been proposed to have pro-malignant properties and regulate cell responses such as proliferation, migration, and inflammation. We used the parental human breast cancer cell line MDA-MB-231 and two single cell progenies derived from lung and bone metastasis upon injection of the parental cells into immuno-deficient mice. The lung and the bone metastatic cell lines showed a marked upregulation of CerK mRNA and activity when compared to the parental cell line. The metastatic cells also had increased migratory and invasive activity, which was dose-dependently reduced by the selective CerK inhibitor NVP-231. -

Inhibition of Spinach Phosphoribulokinase by DL-Glyceraldehyde by ANTONI R
Biochem. J. (1976) 153, 613-619 613 Printed in Great Britain Inhibition of Spinach Phosphoribulokinase by DL-Glyceraldehyde By ANTONI R. SLABAS and DAVID A. WALKER Department ofBotany, University ofSheffield, Sheffield S1O 2TN, U.K. (Received 10 September 1975) Spinach chloroplast phosphoribulokinase is inhibited by DL-glyceraldehyde. The in- hibition is non-competitive with respect to ribulose 5-phosphate (Ki 19mM) and ATP (Ki 20mM). The inhibition is discussed in relation to a previously reported inhibition of CO2 assimilation in intact and envelope-free chloroplasts by DL-glyceraldehyde. It is concluded that the inhibition of phosphoribulokinase is insufficient to account for the inhibition, by DL-glyceraldehyde, of 02 evolution with ribose 5-phosphate as substrate and that a further site of inhibition is also present in this system. DL-Glyceraldehyde inhibits photosynthetic carbon glycylglycine buffer, pH7.4, in a total volume of assiniilation by intact chloroplasts and by the re- 13 ml. The reaction was terminated by the addition constituted chloroplast system (Stokes & Walker, of 2ml of 50 % (w/v) trichloroacetic acid and the pH 1972). The inhibition is most pronounced with intact was adjusted to pH8.0 with KOH. After the addition chloroplasts, a concentration of 1OmM being sufficient of Sml of 1.OM-barium acetate the solution was to suppress 02 evolution completely. It is important centrifuged and the precipitate discarded after because DL-glyceraldehyde is the only known inhibi- washing with Sml of water. Cold ethanol (4vol.) was tor of photosynthesis that is entirely without detect- added to the combined supernatants (total volume able effect on photophosphorylation or photo- 25.4ml); the new precipitate was recovered by synthetic electron transport. -

Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase†,‡ Kap Lim,§ Randy J
Biochemistry 2007, 46, 14845-14853 14845 Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase†,‡ Kap Lim,§ Randy J. Read,| Celia C. H. Chen,§ Aleksandra Tempczyk,§ Min Wei,⊥ Dongmei Ye,⊥ Chun Wu,⊥ Debra Dunaway-Mariano,⊥ and Osnat Herzberg*,§ Center for AdVanced Research in Biotechnology, UniVersity of Maryland Biotechnology Institute, RockVille, Maryland 20850, Department of Haematology, Cambridge Institute for Medical Research, UniVersity of Cambridge, Cambridge, United Kingdom, and Department of Chemistry, UniVersity of New Mexico, Albuquerque, New Mexico ReceiVed September 10, 2007; ReVised Manuscript ReceiVed October 17, 2007 ABSTRACT: Pyruvate phosphate dikinase (PPDK) catalyzes the reversible conversion of phosphoenolpyruvate (PEP), AMP, and Pi to pyruvate and ATP. The enzyme contains two remotely located reaction centers: the nucleotide partial reaction takes place at the N-terminal domain, and the PEP/pyruvate partial reaction takes place at the C-terminal domain. A central domain, tethered to the N- and C-terminal domains by two closely associated linkers, contains a phosphorylatable histidine residue (His455). The molecular architecture suggests a swiveling domain mechanism that shuttles a phosphoryl group between the two reaction centers. In an early structure of PPDK from Clostridium symbiosum, the His445-containing domain (His domain) was positioned close to the nucleotide binding domain and did not contact the PEP/pyruvate- binding domain. Here, we present the crystal structure of a second conformational state of C. symbiosum PPDK with the His domain adjacent to the PEP-binding domain. The structure was obtained by producing a three-residue mutant protein (R219E/E271R/S262D) that introduces repulsion between the His and nucleotide-binding domains but preserves viable interactions with the PEP/pyruvate-binding domain. -

Ceramides: Nutrient Signals That Drive Hepatosteatosis
J Lipid Atheroscler. 2020 Jan;9(1):50-65 Journal of https://doi.org/10.12997/jla.2020.9.1.50 Lipid and pISSN 2287-2892·eISSN 2288-2561 Atherosclerosis Review Ceramides: Nutrient Signals that Drive Hepatosteatosis Scott A. Summers Department of Nutrition and Integrative Physiology, University of Utah, Salt Lake City, UT, USA Received: Sep 24, 2019 ABSTRACT Revised: Nov 4, 2019 Accepted: Nov 10, 2019 Ceramides are minor components of the hepatic lipidome that have major effects on liver Correspondence to function. These products of lipid and protein metabolism accumulate when the energy needs Scott A. Summers of the hepatocyte have been met and its storage capacity is full, such that free fatty acids start Department of Nutrition and Integrative to couple to the sphingoid backbone rather than the glycerol moiety that is the scaffold for Physiology, University of Utah, 15N 2030E, Salt Lake City, UT 84112, USA. glycerolipids (e.g., triglycerides) or the carnitine moiety that shunts them into mitochondria. E-mail: [email protected] As ceramides accrue, they initiate actions that protect cells from acute increases in detergent- like fatty acids; for example, they alter cellular substrate preference from glucose to lipids Copyright © 2020 The Korean Society of Lipid and they enhance triglyceride storage. When prolonged, these ceramide actions cause insulin and Atherosclerosis. This is an Open Access article distributed resistance and hepatic steatosis, 2 of the underlying drivers of cardiometabolic diseases. under the terms of the Creative Commons Herein the author discusses the mechanisms linking ceramides to the development of insulin Attribution Non-Commercial License (https:// resistance, hepatosteatosis and resultant cardiometabolic disorders. -

Glucokinase September 15, 2005
Glucokinase September 15, 2005 Summary Glucokinase phosphorylates glucose into glucose 6-phosphate using ATP as the phosphate group donor, according to the reaction glucose + ATP Æ glucose 6-phosphate + ADP This protocol describes an indirect assay to determine the activity of glucokinase. Glucose 6- phosphate formed by glucokinase is measured by the formation of NADPH in presence of glucose 6-phosphate dehydrogenase. Solutions Required 1. 100 mM tris HCl pH 7.4 Stock solution could be used 2. 100 mM MgSO4·7H2O Stock solution could be used 3. 10 mM glucose Must be prepared fresh 4. 20 mM ATP Must be prepared fresh 5. 10 mM NADP Must be prepared fresh 6. a solution containing 10 U of glucose 6-phosphate dehydrogenase per 50 μL in tris HCl pH 7.4 buffer. This solution is prepared by mixing 40 μL of a 5000 U/mL solution (Sigma G8529) with 960 μL tris buffer. Preparation of Cell Extract Follow general protocol described in Preparation of Cell Extract. The cell extract should be suspended in tris buffer after pelletization. Spectrophotometer Turn on the ultraviolet bulb on the spectrophotometer (Beckman DU50) and wait 10 minutes for warm-up. Select the kinetics-time window on the instrument. Load the method "A:/nadh" ". These methods each have a run-time of 120 s, a temperature of 37°C, a wavelength of 340 nm and use 2 autosamplers. Procedure 1. For each assay, prepare the two cocktails shown in the following table into two separate quartz cuvettes. Keep them on ice. Volume (μL) added to: Solution Control Experimental tris 500 500 DI H2O 200 100 MgSO4 100 100 glucose 0 100 ATP 50 50 NADP 50 50 G6P dehydrogenase 50 50 2. -

Inhibition of Human Telomerase Enhances the Effect of the Tyrosine Kinase Inhibitor, Imatinib, in BCR-ABL-Positive Leukemia Cells1
Vol. 8, 3341–3347, November 2002 Clinical Cancer Research 3341 Advances in Brief Inhibition of Human Telomerase Enhances the Effect of the Tyrosine Kinase Inhibitor, Imatinib, in BCR-ABL-positive Leukemia Cells1 Tetsuzo Tauchi,2 Akihiro Nakajima, by only two amino acids, exhibited normal morphology. The Goro Sashida, Takashi Shimamoto, evidence of apoptosis in the telomerase-inhibited cells was Junko H. Ohyashiki, Kenji Abe, determined by flow cytometric analysis with APO2.7 mono- clonal antibody. We also observed enhanced induction of Kohtaro Yamamoto, and Kazuma Ohyashiki apoptosis by imatinib seen in DN-hTERT-expressing K562 First Department of Internal Medicine, Tokyo Medical University, cells, as compared with WT-hTERT-expressing cells. Tokyo, 160-0023 [T. T., A. N., G. S., T. S., K. O.]; Intractable Conclusions: These results demonstrate that disruption Disease Research Center, Tokyo Medical University, Shinjuku-ku, Tokyo 160-0023 [J. H. O.]; and Department of Virology, Medical of telomere maintenance limits the cellular life span of leu- Research Institute, Tokyo Medical and Dental University, Tokyo, kemia cells and show that the combined use of imatinib and 113-8519 [K. A., K. Y.], Japan telomere maintenance inhibition may be effective in the treatment of BCR-ABL-positive leukemia. Abstract Purpose: Telomerase is a ribonucleoprotein enzyme Introduction that maintains protective structures at the ends of eukary- BCR-ABL is a chimeric oncoprotein generated by recip- otic chromosomes. Earlier findings have supported an rocal translocation between chromosomes 9 and 22 and impli- association between progressive telomere shortening in cated in the pathogenesis of Ph3-positive leukemia (1). BCR- the chronic phase of chronic myelogenous leukemia and the ABL fusion proteins exhibit elevated tyrosine kinase activity up-regulation of telomerase activity occurring late in and transforming properties (2, 3). -

Standard Operating Procedure Procedure in Vitro Kinase Assay
Standard Operating Procedure Procedure In Vitro Kinase Assay with [γ-32P] - ATP Department Location SOP Prepared By: Section 1: Purpose Kinase assays are used to detect the activity of specific kinases from cells. Kinases are a group of enzymes that modifies a target substrate by phosphorylation. In this assay, the kinase removes the radioactively labeled phosphate group from [γ-32P] - ATP and adds it to the target substrate. Kinase activity is measured by running an SDS PAGE (gel electrophoresis for proteins) and visualizing the results with an imaging technique which quantifies the amount of radiolabeled substrate. Given kinases’ essential role in cell metabolism, signaling, etc, this assay can be applied virtually to any biological research that wishes to investigate enzymatic activity within a cellular process. Some studies include measuring yeast kinases to understand chromosomal segregation1 and measuring protein kinase R (PKR) activity2, an important modulator of chronic metabolic inflammation associated to obesity, diabetes, and cardiovascular disease. Section 2: Personal Protective Equipment and Survey Equipment PPE: • Lab coat • Nitrile Gloves • Heat resistant gloves • Safety Glasses • Closed-toe shoes Other Equipment: • Geiger counter with pancake probe • Personal chest dosimeter • Personal finger dosimeter Section 3: Radioactive Material ATP, [γ-32P], Supplier: Perkin Elmer Starting activity: 10 uCi/uL Typical use quantities: 100 uCi - 1mCi per experiment • Radioactive material exposure (RAM) time on benchtop: ~30 minutes Activity used per experiment: _______________ RAM handling time: _______________ Frequency of experiment: _______________ Section 4: Potential Hazards • 32P is a high-energy beta emitter and has a half-life of 14.29 days. 32P can present a substantial skin and eye dose hazard. -

Role of Ceramide Kinase in Breast Cancer Progression
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2014 Role of Ceramide Kinase in Breast Cancer Progression Ania Warczyk Payne University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Cell Biology Commons, Molecular Biology Commons, and the Pharmacology Commons Recommended Citation Payne, Ania Warczyk, "Role of Ceramide Kinase in Breast Cancer Progression" (2014). Publicly Accessible Penn Dissertations. 1402. https://repository.upenn.edu/edissertations/1402 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1402 For more information, please contact [email protected]. Role of Ceramide Kinase in Breast Cancer Progression Abstract Recurrent breast cancer is typically an incurable disease and, as such, is disproportionately responsible for deaths from this disease. Recurrent breast cancers arise from the pool of disseminated tumor cells (DTCs) that survive adjuvant or neoadjuvant therapy, and patients with detectable DTCs following therapy are at substantially increased risk for recurrence. Consequently, the identification of pathways that contribute to the survival of breast cancer cells following therapy could aid in the development of more effective therapies that decrease the burden of residual disease and thereby reduce the risk of breast cancer recurrence. We now report that Ceramide Kinase (Cerk) is required for mammary tumor recurrence following HER2/neu pathway inhibition and is spontaneously up-regulated during tumor recurrence in multiple genetically engineered mouse models for breast cancer. We find that Cerk is apidlyr up-regulated in tumor cells following HER2/neu down-regulation or treatment with adriamycin and that Cerk is required for tumor cell survival following HER2/neu down-regulation. -

Pyruvate Orthophosphate Dikinase
Chapter 15 Structure, Function, and Post-translational Regulation of C4 Pyruvate Orthophosphate Dikinase Chris J. Chastain Department of Biosciences, Minnesota State University-Moorhead , Moorhead , MN 56563 , USA Summary .............................................................................................................................................................. 301 I. Introduction .................................................................................................................................................... 302 A. Role of PPDK in C 4 Plants ...................................................................................................................... 302 B. PPDK Enzyme Properties ...................................................................................................................... 302 1. Catalysis as Related to Structure ..................................................................................................... 302 2. Oligomeric Structure and Tetramer Dissociation at Cool Temperatures ........................................... 304 3. Substrate Km s for C 4 PPDK .............................................................................................................. 304 C. PPDK as a Rate-Limiting Enzyme of the C 4 Pathway ............................................................................ 304 II. Post-translational Regulation of C 4 PPDK ..................................................................................................... 305 A. Light/Dark -

The Serine/Threonine Protein Kinase (Akt)/ Protein Kinase B (Pkb) Signaling Pathway in Breast Cancer
Journal of Mind and Medical Sciences Volume 7 Issue 1 Article 7 2020 The Serine/Threonine Protein Kinase (Akt)/ Protein Kinase B (PkB) Signaling Pathway in Breast Cancer Daniela Miricescu FACULTY OF DENTAL MEDICINE, DEPARTMENT OF BIOCHEMISTRY, BUCHAREST, ROMANIA Camelia Cristina Diaconu CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, CLINICAL EMERGENCY HOSPITAL OF BUCHAREST, INTERNAL MEDICINE CLINIC, BUCHAREST, ROMANIA Constantin Stefani CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, DEPARTMENT OF FAMILY MEDICINE, BUCHAREST, ROMANIA Ana Maria Alexandra Stanescu CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, DEPARTMENT OF FAMILY MEDICINE, BUCHAREST, ROMANIA Alexandra Totan FAollowCULT thisY OF and DEN additionalTAL MEDICINE, works at:DEP https:/ARTMEN/scholarT OF.v BIOCHEMISTRalpo.edu/jmmsY, BUCHAREST, ROMANIA Part of the Obstetrics and Gynecology Commons, Oncology Commons, and the Palliative Care SeeCommons next page for additional authors Recommended Citation Miricescu, Daniela; Diaconu, Camelia Cristina; Stefani, Constantin; Stanescu, Ana Maria Alexandra; Totan, Alexandra; Rusu, Ioana Ruxandra; Bratu, Ovidiu Gabriel; Spinu, Dan; and Greabu, Maria (2020) "The Serine/ Threonine Protein Kinase (Akt)/ Protein Kinase B (PkB) Signaling Pathway in Breast Cancer," Journal of Mind and Medical Sciences: Vol. 7 : Iss. 1 , Article 7. DOI: 10.22543/7674.71.P3439 Available at: https://scholar.valpo.edu/jmms/vol7/iss1/7 This Review Article is brought to you for free and open access by ValpoScholar. It has been accepted for inclusion in Journal -

Differential Regulation of Telomeric Complex by BCR-ABL1 Kinase In
G C A T T A C G G C A T genes Article Differential Regulation of Telomeric Complex by BCR-ABL1 Kinase in Human Cellular Models of Chronic Myeloid Leukemia—From Single Cell Analysis to Next-Generation Sequencing 1,2, 2,3, 3 2,3 Anna Deregowska y, Monika Pepek y, Katarzyna Pruszczyk , Marcin M. Machnicki , Maciej Wnuk 1,* and Tomasz Stoklosa 4,* 1 Department of Biotechnology, University of Rzeszow, Pigonia 1, 35-310 Rzeszów, Poland; [email protected] 2 Postgraduate School of Molecular Medicine, Medical University of Warsaw, Trojdena 2a, 02-091 Warsaw, Poland; [email protected] (M.P.); [email protected] (M.M.M.) 3 Department of Immunology, Medical University of Warsaw, Nielubowicza 5, 02-097 Warsaw, Poland; [email protected] 4 Department of Tumor Biology and Genetics, Medical University of Warsaw, Pawinskiego 7, 02-106 Warsaw, Poland * Correspondence: [email protected] (M.W.); [email protected] (T.S.); Tel.: +4-822-599-2199 (T.S.) These authors contributed equally to this work. y Received: 8 August 2020; Accepted: 25 September 2020; Published: 29 September 2020 Abstract: Telomeres are specialized nucleoprotein complexes, localized at the physical ends of chromosomes, that contribute to the maintenance of genome stability. One of the features of chronic myeloid leukemia (CML) cells is a reduction in telomere length which may result in increased genomic instability and progression of the disease. Aberrant telomere maintenance in CML is not fully understood and other mechanisms such as the alternative lengthening of telomeres (ALT) are involved. In this work, we employed five BCR-ABL1-positive cell lines, namely K562, KU-812, LAMA-84, MEG-A2, and MOLM-1, commonly used in the laboratories to study the link between mutation, copy number, and expression of telomere maintenance genes with the expression, copy number, and activity of BCR-ABL1.