Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
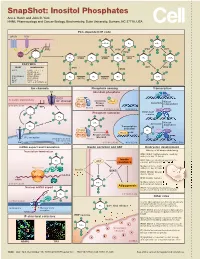
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

Ceramide Metabolism Regulates a Neuronal Nadph Oxidase Influencing Neuron Survival During Inflammation
Ceramide Metabolism Regulates A Neuronal Nadph Oxidase Influencing Neuron Survival During Inflammation Item Type Thesis Authors Barth, Brian M. Download date 07/10/2021 02:29:56 Link to Item http://hdl.handle.net/11122/8999 CERAMIDE METABOLISM REGULATES A NEURONAL NADPH OXIDASE INFLUENCING NEURON SURVIVAL DURING INFLAMMATION A THESIS Presented to the Faculty of the University of Alaska Fairbanks in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY By Brian M. Barth, B.S., M.S. Fairbanks, Alaska August 2009 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. UMI Number: 3386045 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI Dissertation Publishing UMI 3386045 Copyright 2009 by ProQuest LLC. All rights reserved. This edition of the work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. CERAMIDE METABOLISM REGULATES A NEURONAL NADPH OXIDASE INFLUENCING NEURON SURVIVAL DURING INFLAMMATION By Brian M. Barth RECOMMENDED: , ... f / M Advisory Committee Chah Chai ^Department of Chemistry and Biochemistry /? & f ) ./ APPROVED: Dean, C olleg£^ Natural Science'and“Mathemati cs /<C £>dan of the Graduate School 3 / / - Date Reproduced with permission of the copyright owner. -

When a Slight Tilt Is Enough
Cell. Mol. Life Sci. (2013) 70:181–203 DOI 10.1007/s00018-012-1038-x Cellular and Molecular Life Sciences REVIEW Ceramide function in the brain: when a slight tilt is enough Chiara Mencarelli • Pilar Martinez–Martinez Received: 11 January 2012 / Revised: 16 May 2012 / Accepted: 21 May 2012 / Published online: 24 June 2012 Ó The Author(s) 2012. This article is published with open access at Springerlink.com Abstract Ceramide, the precursor of all complex sphin- Introduction golipids, is a potent signaling molecule that mediates key events of cellular pathophysiology. In the nervous system, Ceramides are a family of lipid molecules that consist of the sphingolipid metabolism has an important impact. sphingoid long-chain base linked to an acyl chain via an Neurons are polarized cells and their normal functions, amide bond. Ceramides differ from each other by length, such as neuronal connectivity and synaptic transmission, hydroxylation, and saturation of both the sphingoid base rely on selective trafficking of molecules across plasma and fatty acid moieties. membrane. Sphingolipids are abundant on neural cellular Sphingoid bases are of three general chemical types: membranes and represent potent regulators of brain sphingosine, dihydrosphingosine (commonly known as homeostasis. Ceramide intracellular levels are fine-tuned ‘‘sphinganine’’, as it will be addressed in this review) and and alteration of the sphingolipid–ceramide profile con- phytosphingosine. Based on the nature of the sphingoid tributes to the development of age-related, neurological base backbone, we can distinguish three main subgroups in and neuroinflammatory diseases. The purpose of this the ceramide family: the compound named ceramide con- review is to guide the reader towards a better understanding tains sphingosine, which has a trans-double bond at the of the sphingolipid–ceramide pathway system. -

Suppression of Mast Cell Degranulation by a Novel Ceramide Kinase Inhibitor, the F-12509A Olefin Isomer K1
Title Suppression of mast cell degranulation by a novel ceramide kinase inhibitor, the F-12509A olefin isomer K1 Kim, Jin-Wook; Inagaki, Yuichi; Mitsutake, Susumu; Maezawa, Nobuhiro; Katsumura, Shigeo; Ryu, Yeon-Woo; Park, Author(s) Chang-Seo; Taniguchi, Masaru; Igarashi, Yasuyuki Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1738(1-3), 82-90 Citation https://doi.org/10.1016/j.bbalip.2005.10.007 Issue Date 2005 Doc URL http://hdl.handle.net/2115/5800 Type article (author version) File Information BBA1738(1-3).pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP 1 Suppression of mast cell degranulation by a novel ceramide kinase inhibitor, the F-12509A olefin isomer K1 Jin-Wook Kima, b, c, 1, Yuichi Inagakia, 1, Susumu Mitsutakea, Nobuhiro Maezawad, Shigeo Katsumurad, Yeon-Woo Ryub, Chang-Seo Parke, Masaru Taniguchif, and Yasuyuki Igarashia aDepartment of Biomembrane and Biofunctional Chemistry, Graduate School of Pharmaceutical Science, Hokkaido University. Kita 12, Nishi 6, Kita-ku, Sapporo 060-0812, Japan, bDepartment of Molecular Science and Technology, Ajou University, San 5, Wonchun-dong, Yeongtong-gu, Suwon 443-749, Korea, cDoosan Biotech, 39-3, Seongbok-dong, Yongin-si, Gyeonggi-do 449-795, Korea, dSchool of Science and Technology, Kwansei Gakuin University, Gakuen, Sanda, Hyogo 669-1337, Japan, eDepartment of Chemical and Biochemical Engineering, Dongguk University, 3-26 Pil-dong, Chung-gu, Seoul 100-715, Korea, fRIKEN Research Center for Allergy and Immunology, Yokohama, Kanagawa 230-0045, Japan Key words: ceramide, ceramide 1-phosphate, ceramide kinase, inhibitor, degranulation, mast cell To whom correspondence should be addressed: Dr. -

Inhibition of Spinach Phosphoribulokinase by DL-Glyceraldehyde by ANTONI R
Biochem. J. (1976) 153, 613-619 613 Printed in Great Britain Inhibition of Spinach Phosphoribulokinase by DL-Glyceraldehyde By ANTONI R. SLABAS and DAVID A. WALKER Department ofBotany, University ofSheffield, Sheffield S1O 2TN, U.K. (Received 10 September 1975) Spinach chloroplast phosphoribulokinase is inhibited by DL-glyceraldehyde. The in- hibition is non-competitive with respect to ribulose 5-phosphate (Ki 19mM) and ATP (Ki 20mM). The inhibition is discussed in relation to a previously reported inhibition of CO2 assimilation in intact and envelope-free chloroplasts by DL-glyceraldehyde. It is concluded that the inhibition of phosphoribulokinase is insufficient to account for the inhibition, by DL-glyceraldehyde, of 02 evolution with ribose 5-phosphate as substrate and that a further site of inhibition is also present in this system. DL-Glyceraldehyde inhibits photosynthetic carbon glycylglycine buffer, pH7.4, in a total volume of assiniilation by intact chloroplasts and by the re- 13 ml. The reaction was terminated by the addition constituted chloroplast system (Stokes & Walker, of 2ml of 50 % (w/v) trichloroacetic acid and the pH 1972). The inhibition is most pronounced with intact was adjusted to pH8.0 with KOH. After the addition chloroplasts, a concentration of 1OmM being sufficient of Sml of 1.OM-barium acetate the solution was to suppress 02 evolution completely. It is important centrifuged and the precipitate discarded after because DL-glyceraldehyde is the only known inhibi- washing with Sml of water. Cold ethanol (4vol.) was tor of photosynthesis that is entirely without detect- added to the combined supernatants (total volume able effect on photophosphorylation or photo- 25.4ml); the new precipitate was recovered by synthetic electron transport. -

Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase†,‡ Kap Lim,§ Randy J
Biochemistry 2007, 46, 14845-14853 14845 Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase†,‡ Kap Lim,§ Randy J. Read,| Celia C. H. Chen,§ Aleksandra Tempczyk,§ Min Wei,⊥ Dongmei Ye,⊥ Chun Wu,⊥ Debra Dunaway-Mariano,⊥ and Osnat Herzberg*,§ Center for AdVanced Research in Biotechnology, UniVersity of Maryland Biotechnology Institute, RockVille, Maryland 20850, Department of Haematology, Cambridge Institute for Medical Research, UniVersity of Cambridge, Cambridge, United Kingdom, and Department of Chemistry, UniVersity of New Mexico, Albuquerque, New Mexico ReceiVed September 10, 2007; ReVised Manuscript ReceiVed October 17, 2007 ABSTRACT: Pyruvate phosphate dikinase (PPDK) catalyzes the reversible conversion of phosphoenolpyruvate (PEP), AMP, and Pi to pyruvate and ATP. The enzyme contains two remotely located reaction centers: the nucleotide partial reaction takes place at the N-terminal domain, and the PEP/pyruvate partial reaction takes place at the C-terminal domain. A central domain, tethered to the N- and C-terminal domains by two closely associated linkers, contains a phosphorylatable histidine residue (His455). The molecular architecture suggests a swiveling domain mechanism that shuttles a phosphoryl group between the two reaction centers. In an early structure of PPDK from Clostridium symbiosum, the His445-containing domain (His domain) was positioned close to the nucleotide binding domain and did not contact the PEP/pyruvate- binding domain. Here, we present the crystal structure of a second conformational state of C. symbiosum PPDK with the His domain adjacent to the PEP-binding domain. The structure was obtained by producing a three-residue mutant protein (R219E/E271R/S262D) that introduces repulsion between the His and nucleotide-binding domains but preserves viable interactions with the PEP/pyruvate-binding domain. -

Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy
cancers Review Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy Preeti Gupta 1, Aaliya Taiyab 1 , Afzal Hussain 2, Mohamed F. Alajmi 2, Asimul Islam 1 and Md. Imtaiyaz Hassan 1,* 1 Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India; [email protected] (P.G.); [email protected] (A.T.); [email protected] (A.I.) 2 Department of Pharmacognosy, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia; afi[email protected] (A.H.); [email protected] (M.F.A.) * Correspondence: [email protected] Simple Summary: Cancer is the prime cause of death globally. The altered stimulation of signaling pathways controlled by human kinases has often been observed in various human malignancies. The over-expression of SphK1 (a lipid kinase) and its metabolite S1P have been observed in various types of cancer and metabolic disorders, making it a potential therapeutic target. Here, we discuss the sphingolipid metabolism along with the critical enzymes involved in the pathway. The review provides comprehensive details of SphK isoforms, including their functional role, activation, and involvement in various human malignancies. An overview of different SphK inhibitors at different phases of clinical trials and can potentially be utilized as cancer therapeutics has also been reviewed. Citation: Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Abstract: Sphingolipid metabolites have emerged as critical players in the regulation of various Hassan, M..I. Targeting the Sphingosine Kinase/Sphingosine- physiological processes. Ceramide and sphingosine induce cell growth arrest and apoptosis, whereas 1-Phosphate Signaling Axis in Drug sphingosine-1-phosphate (S1P) promotes cell proliferation and survival. -

Ceramides: Nutrient Signals That Drive Hepatosteatosis
J Lipid Atheroscler. 2020 Jan;9(1):50-65 Journal of https://doi.org/10.12997/jla.2020.9.1.50 Lipid and pISSN 2287-2892·eISSN 2288-2561 Atherosclerosis Review Ceramides: Nutrient Signals that Drive Hepatosteatosis Scott A. Summers Department of Nutrition and Integrative Physiology, University of Utah, Salt Lake City, UT, USA Received: Sep 24, 2019 ABSTRACT Revised: Nov 4, 2019 Accepted: Nov 10, 2019 Ceramides are minor components of the hepatic lipidome that have major effects on liver Correspondence to function. These products of lipid and protein metabolism accumulate when the energy needs Scott A. Summers of the hepatocyte have been met and its storage capacity is full, such that free fatty acids start Department of Nutrition and Integrative to couple to the sphingoid backbone rather than the glycerol moiety that is the scaffold for Physiology, University of Utah, 15N 2030E, Salt Lake City, UT 84112, USA. glycerolipids (e.g., triglycerides) or the carnitine moiety that shunts them into mitochondria. E-mail: [email protected] As ceramides accrue, they initiate actions that protect cells from acute increases in detergent- like fatty acids; for example, they alter cellular substrate preference from glucose to lipids Copyright © 2020 The Korean Society of Lipid and they enhance triglyceride storage. When prolonged, these ceramide actions cause insulin and Atherosclerosis. This is an Open Access article distributed resistance and hepatic steatosis, 2 of the underlying drivers of cardiometabolic diseases. under the terms of the Creative Commons Herein the author discusses the mechanisms linking ceramides to the development of insulin Attribution Non-Commercial License (https:// resistance, hepatosteatosis and resultant cardiometabolic disorders. -

Ceramide Kinase and Ceramide-1-Phosphate
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2008 Ceramide Kinase and Ceramide-1-Phosphate Dayanjan Wijesinghe Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Biochemistry, Biophysics, and Structural Biology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/1621 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Dayanjan S Wijesinghe 2008 All Rights Reserved CERAMIDE KINASE AND CERAMIDE 1 PHOSPHATE A Dissertation submitted in partial fulfillment of the requirements for the degree of PhD at Virginia Commonwealth University. by DAYANJAN SHANAKA WIJESINGHE BSc., University of Peradeniya, Sri Lanka, 2001 Grad. I. Chem. C., Institute of Chemistry, Sri Lanka, 1998 Director: CHARLES E. CHALFANT ASSISTANT PROFFESSOR DEPARTMENT OF BIOCHEMISTRY AND MOLECULAR BIOLOGY Virginia Commonwealth University Richmond, Virginia December 2008 ii DEDICATION To my darling wife Piumini, my daughter Nisha, my Mother, Father and my Father and Mother-in-Law for their unconditional love and support. iii Acknowledgement I would like to express my heartfelt gratitude to my adviser, Dr. Charles E Chalfant for his guidance, counsel and advice without towards successful completion of this thesis. I am thankful to the members of my committee: Dr. Sarah Spiegel, Dr. Darrell L Peterson, Dr. Stephen T Sawyer and Dr. Lynne Elmore, for their valuable suggestions and time towards the successful completion of my degree. -

Glucokinase September 15, 2005
Glucokinase September 15, 2005 Summary Glucokinase phosphorylates glucose into glucose 6-phosphate using ATP as the phosphate group donor, according to the reaction glucose + ATP Æ glucose 6-phosphate + ADP This protocol describes an indirect assay to determine the activity of glucokinase. Glucose 6- phosphate formed by glucokinase is measured by the formation of NADPH in presence of glucose 6-phosphate dehydrogenase. Solutions Required 1. 100 mM tris HCl pH 7.4 Stock solution could be used 2. 100 mM MgSO4·7H2O Stock solution could be used 3. 10 mM glucose Must be prepared fresh 4. 20 mM ATP Must be prepared fresh 5. 10 mM NADP Must be prepared fresh 6. a solution containing 10 U of glucose 6-phosphate dehydrogenase per 50 μL in tris HCl pH 7.4 buffer. This solution is prepared by mixing 40 μL of a 5000 U/mL solution (Sigma G8529) with 960 μL tris buffer. Preparation of Cell Extract Follow general protocol described in Preparation of Cell Extract. The cell extract should be suspended in tris buffer after pelletization. Spectrophotometer Turn on the ultraviolet bulb on the spectrophotometer (Beckman DU50) and wait 10 minutes for warm-up. Select the kinetics-time window on the instrument. Load the method "A:/nadh" ". These methods each have a run-time of 120 s, a temperature of 37°C, a wavelength of 340 nm and use 2 autosamplers. Procedure 1. For each assay, prepare the two cocktails shown in the following table into two separate quartz cuvettes. Keep them on ice. Volume (μL) added to: Solution Control Experimental tris 500 500 DI H2O 200 100 MgSO4 100 100 glucose 0 100 ATP 50 50 NADP 50 50 G6P dehydrogenase 50 50 2. -

Inhibition of Human Telomerase Enhances the Effect of the Tyrosine Kinase Inhibitor, Imatinib, in BCR-ABL-Positive Leukemia Cells1
Vol. 8, 3341–3347, November 2002 Clinical Cancer Research 3341 Advances in Brief Inhibition of Human Telomerase Enhances the Effect of the Tyrosine Kinase Inhibitor, Imatinib, in BCR-ABL-positive Leukemia Cells1 Tetsuzo Tauchi,2 Akihiro Nakajima, by only two amino acids, exhibited normal morphology. The Goro Sashida, Takashi Shimamoto, evidence of apoptosis in the telomerase-inhibited cells was Junko H. Ohyashiki, Kenji Abe, determined by flow cytometric analysis with APO2.7 mono- clonal antibody. We also observed enhanced induction of Kohtaro Yamamoto, and Kazuma Ohyashiki apoptosis by imatinib seen in DN-hTERT-expressing K562 First Department of Internal Medicine, Tokyo Medical University, cells, as compared with WT-hTERT-expressing cells. Tokyo, 160-0023 [T. T., A. N., G. S., T. S., K. O.]; Intractable Conclusions: These results demonstrate that disruption Disease Research Center, Tokyo Medical University, Shinjuku-ku, Tokyo 160-0023 [J. H. O.]; and Department of Virology, Medical of telomere maintenance limits the cellular life span of leu- Research Institute, Tokyo Medical and Dental University, Tokyo, kemia cells and show that the combined use of imatinib and 113-8519 [K. A., K. Y.], Japan telomere maintenance inhibition may be effective in the treatment of BCR-ABL-positive leukemia. Abstract Purpose: Telomerase is a ribonucleoprotein enzyme Introduction that maintains protective structures at the ends of eukary- BCR-ABL is a chimeric oncoprotein generated by recip- otic chromosomes. Earlier findings have supported an rocal translocation between chromosomes 9 and 22 and impli- association between progressive telomere shortening in cated in the pathogenesis of Ph3-positive leukemia (1). BCR- the chronic phase of chronic myelogenous leukemia and the ABL fusion proteins exhibit elevated tyrosine kinase activity up-regulation of telomerase activity occurring late in and transforming properties (2, 3). -

Generation of Sphingosine-1-Phosphate Is Enhanced in Biliary Tract Cancer Patients and Is Associated with Lymphatic Metastasis
www.nature.com/scientificreports OPEN Generation of sphingosine- 1-phosphate is enhanced in biliary tract cancer patients and Received: 5 April 2018 Accepted: 4 July 2018 is associated with lymphatic Published: xx xx xxxx metastasis Yuki Hirose1, Masayuki Nagahashi1, Eriko Katsuta2, Kizuki Yuza1, Kohei Miura1, Jun Sakata1, Takashi Kobayashi1, Hiroshi Ichikawa1, Yoshifumi Shimada1, Hitoshi Kameyama1, Kerry-Ann McDonald2, Kazuaki Takabe 1,2,3,4,5 & Toshifumi Wakai1 Lymphatic metastasis is known to contribute to worse prognosis of biliary tract cancer (BTC). Recently, sphingosine-1-phosphate (S1P), a bioactive lipid mediator generated by sphingosine kinase 1 (SPHK1), has been shown to play an important role in lymphangiogenesis and lymph node metastasis in several types of cancer. However, the role of the lipid mediator in BTC has never been examined. Here we found that S1P is elevated in BTC with the activation of ceramide-synthetic pathways, suggesting that BTC utilizes SPHK1 to promote lymphatic metastasis. We found that S1P, sphingosine and ceramide precursors such as monohexosyl-ceramide and sphingomyelin, but not ceramide, were signifcantly increased in BTC compared to normal biliary tract tissue using LC-ESI-MS/MS. Utilizing The Cancer Genome Atlas cohort, we demonstrated that S1P in BTC is generated via de novo pathway and exported via ABCC1. Further, we found that SPHK1 expression positively correlated with factors related to lymphatic metastasis in BTC. Finally, immunohistochemical examination revealed that gallbladder cancer with lymph node metastasis had signifcantly higher expression of phospho-SPHK1 than that without. Taken together, our data suggest that S1P generated in BTC contributes to lymphatic metastasis. Biliary tract cancer (BTC), the malignancy of the bile ducts and gallbladder, is a highly lethal disease in which a strong prognostic predictor is lymph node metastasis1–5.